

This is a preprint.
Replaying germinal center evolution on a quantified affinity landscape
- PMID: 40661619
- PMCID: PMC12258878
- DOI: 10.1101/2025.06.02.656870
Replaying germinal center evolution on a quantified affinity landscape
Abstract
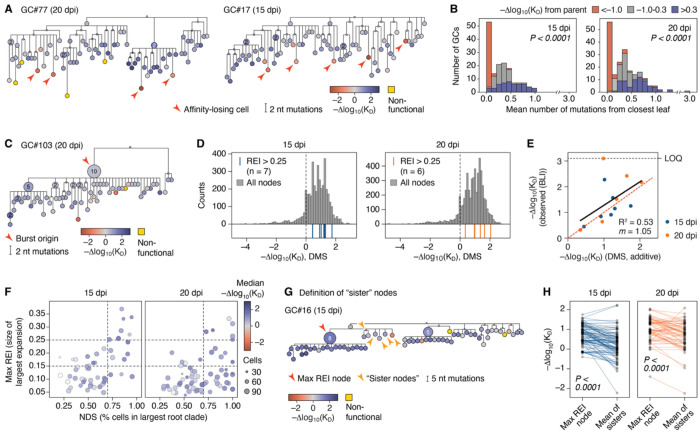
Darwinian evolution of immunoglobulin genes within germinal centers (GC) underlies the progressive increase in antibody affinity following antigen exposure. Whereas the mechanics of how competition between GC B cells drives increased affinity are well established, the dynamical evolutionary features of this process remain poorly characterized. We devised an experimental evolution model in which we "replay" over one hundred instances of a clonally homogenous GC reaction and follow the selective process by assigning affinities to all cells using deep mutational scanning. Our data reveal how GCs achieve predictable evolutionary outcomes through the cumulative effects of many rounds of imperfect selection, acting on a landscape shaped heavily by somatic hypermutation (SHM) targeting biases. Using time-calibrated models, we show that apparent features of GC evolution such as permissiveness to low-affinity lineages and early plateauing of affinity are best explained by survivorship biases that distort our view of how affinity progresses over time.
Conflict of interest statement
Competing interests: G.D.V. and J.D.B. are advisors for and hold stock of the Vaccine Company. J.D.B. consults or has recently consulted for Apriori Bio, Pfizer, and Invivyd on topics related to viruses, vaccines and viral evolution. T.Ara. is currently an employee of Pfizer Inc.
Figures





References
-
- MacLennan I.C. (1994). Germinal centers. Annu Rev Immunol 12, 117–139. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous