

This is a preprint.
ProGuide: a flexible framework for modeling global conformational rearrangements in proteins using DEER-derived distance restraints
- PMID: 40661650
- PMCID: PMC12258946
- DOI: 10.1101/2025.06.05.658127
ProGuide: a flexible framework for modeling global conformational rearrangements in proteins using DEER-derived distance restraints
Abstract
Conformational heterogeneity is integral to protein function - ranging from enzyme catalysis to signal transduction - and visualizing distinct conformational states requires experimental techniques capable of providing such structural information. One particularly powerful method, double electron-electron resonance (DEER) spectroscopy, can provide a high-resolution, long-range (~15-80 Å) probability distributions of distances between site-selected pairs of spin labels to resolve intra-protein distance parameters of unique protein conformations, as well as their respective likelihoods within a conformational ensemble. A current frontier in the field of DEER spectroscopy is utilizing this distance information in computational modeling to generate complete structural models of these multiple conformations. Although several methods have been developed for this purpose, modeling protein backbone structural rearrangements using multiple distance restraints remains challenging, due in part to the complexity provided by rotameric flexibility of the spin label side chain. Here, we overcome these challenges with ProGuide, a new framework for generating accurate structural models guided by DEER distance distribution information. Large conformational rearrangements are captured by performing iterative experimentally biased molecular dynamics simulations. In each iteration, spin-label rotameric heterogeneity is modeled using chiLife, and then changes are calculated to capture distance-probability density present in the experimental DEER distributions and lacking from the modeled one. The resulting models of this process then go through a selection to generate the ensemble that best recapitulates the DEER data. We illustrate the power of this method using published DEER data from a study of biased agonism in the angiotensin II type 1 receptor (AT1R), a prototypical G protein coupled receptor (GPCR). The resulting AT1R models consist of both Gq- and β-arrestin-biased conformations, including a completely novel β-arrestin-biased conformation. These models reveal structural insights involving tertiary structural rearrangements as well as residue-level changes in crucial microswitch motifs. Taken together, the results demonstrate the power and flexibility of ProGuide to investigate conformational rearrangements of large, complex proteins using DEER-derived distance restraints.
Figures
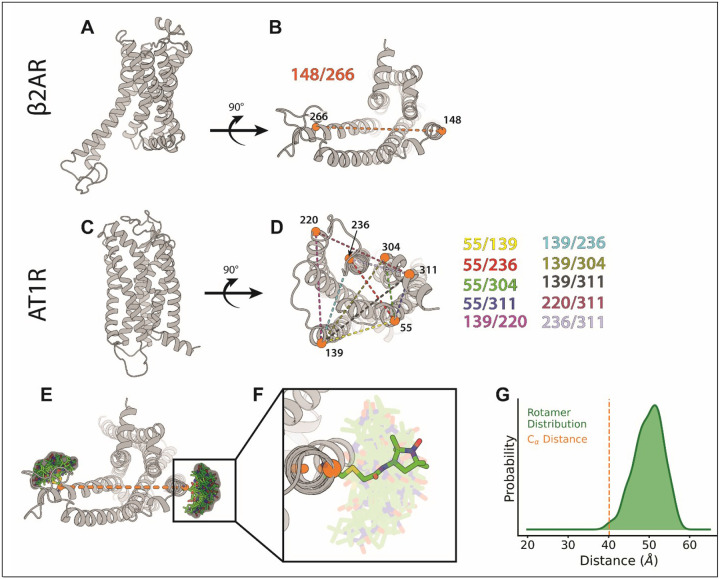
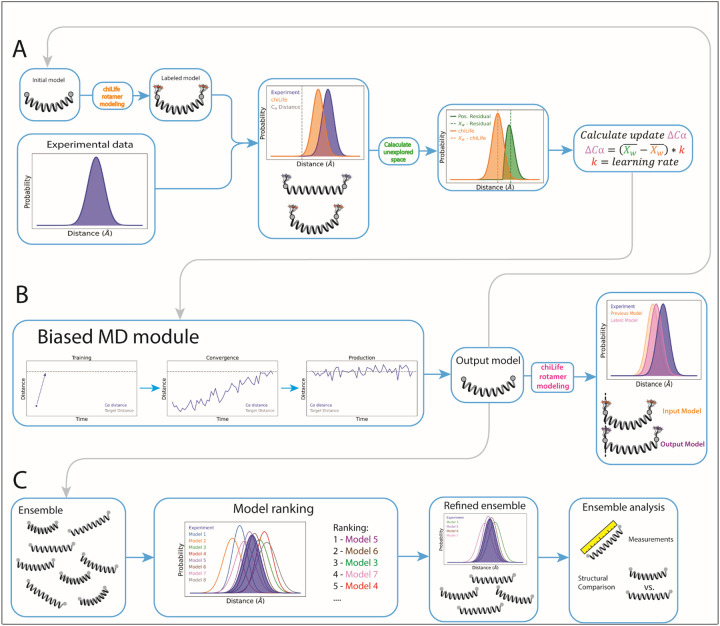
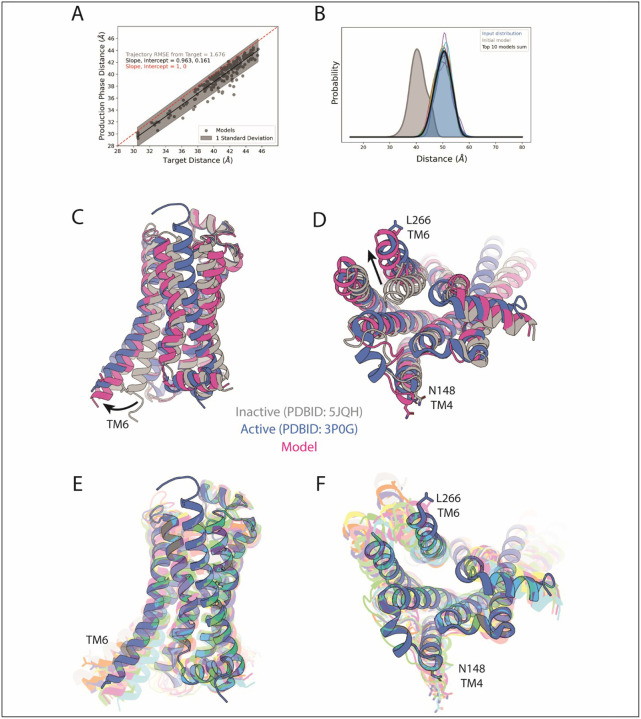
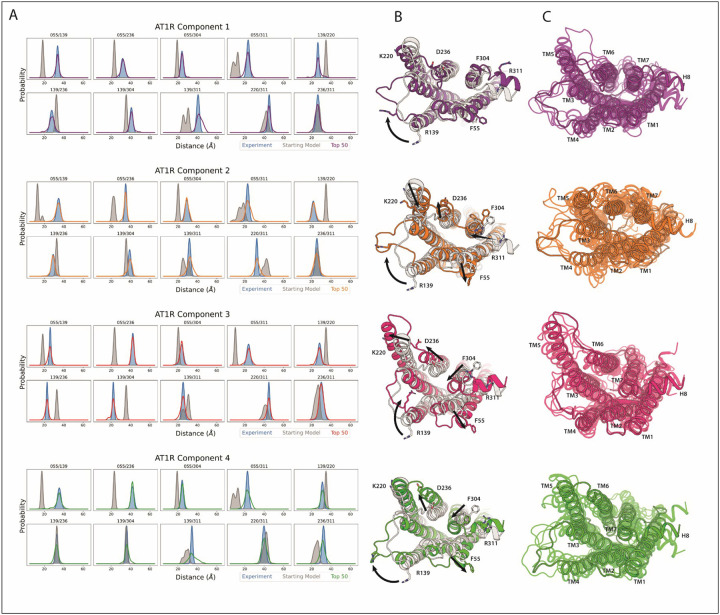

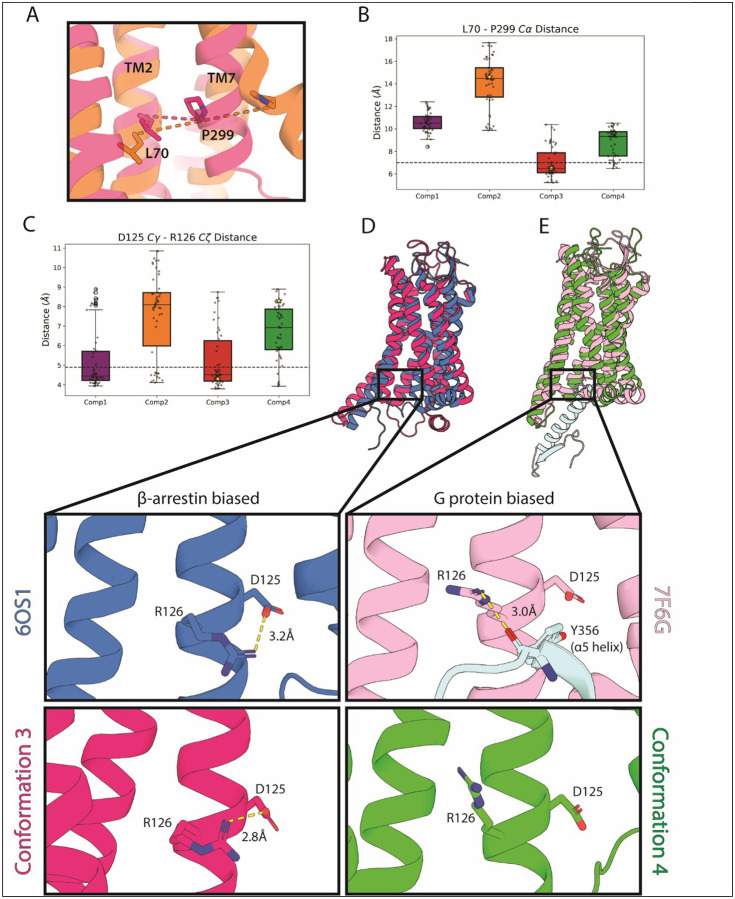
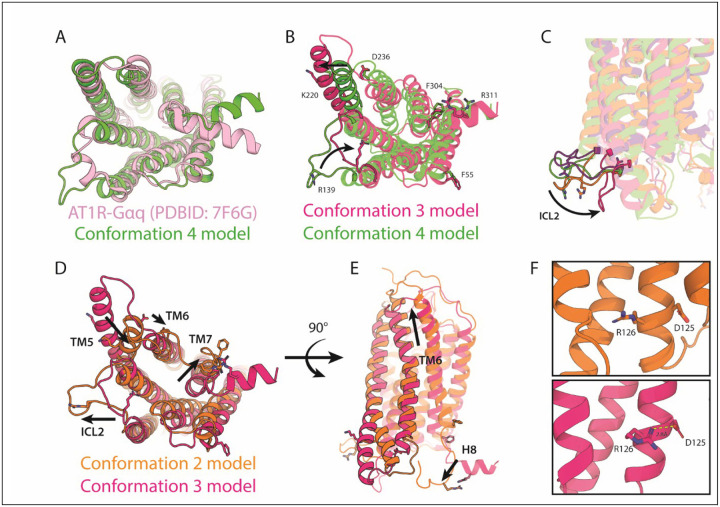
Similar articles
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Electrophoresis.2025 Jul 14. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 14. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 36251838 Free Books & Documents.
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
-
Assessing the comparative effects of interventions in COPD: a tutorial on network meta-analysis for clinicians.Respir Res. 2024 Dec 21;25(1):438. doi: 10.1186/s12931-024-03056-x. Respir Res. 2024. PMID: 39709425 Free PMC article. Review.
References
-
- Frauenfelder H, Sligar SG, Wolynes PG. The Energy Landscapes and Motions of Proteins. Science (1979). 1991;254:1598–1603. https://www.science.org - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources