

Galaxy QCxMS for straightforward semi-empirical quantum mechanical EI-MS prediction
- PMID: 40661852
- PMCID: PMC12257954
- DOI: 10.46471/gigabyte.160
Galaxy QCxMS for straightforward semi-empirical quantum mechanical EI-MS prediction
Abstract
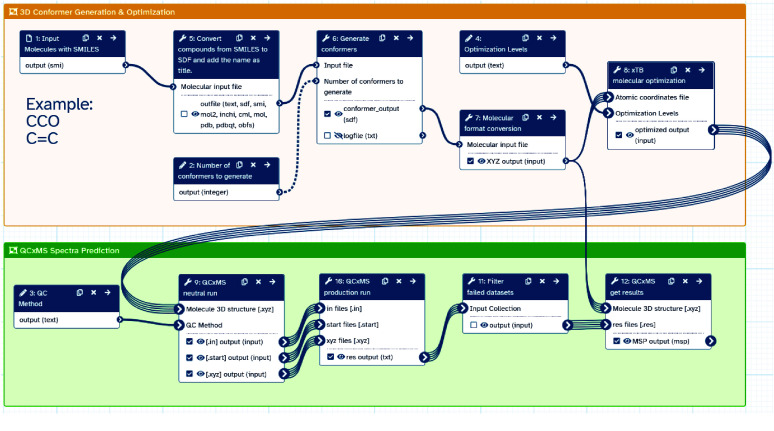
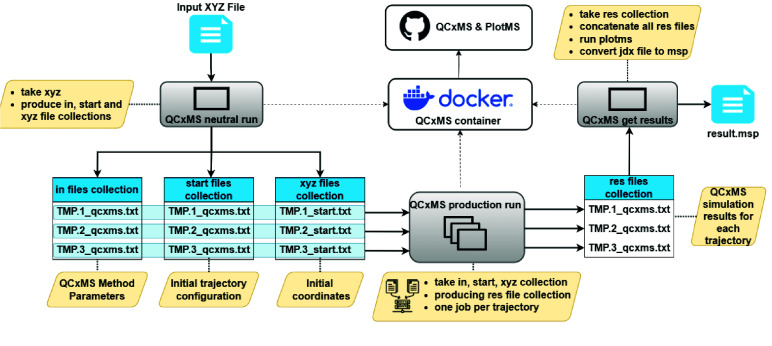
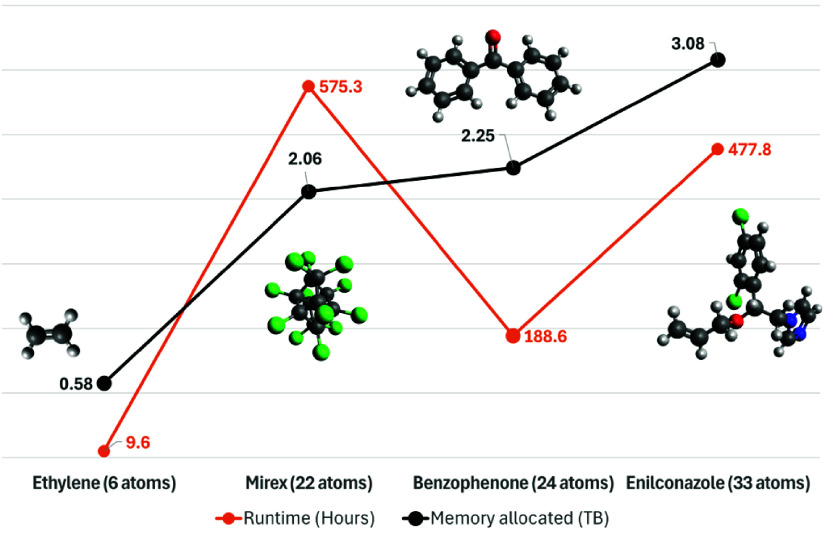
High-performance computing (HPC) environments are crucial for computational research, including quantum chemistry (QC), but pose challenges for non-expert users. Researchers with limited computational knowledge struggle to utilise domain-specific software and access mass spectra prediction for in silico annotation. Here, we provide a robust workflow that leverages interoperable file formats for molecular structures to ensure integration across various QC tools. The quantum chemistry package for mass spectral predictions after electron ionization or collision-induced dissociation has been integrated into the Galaxy platform, enabling automated analysis of fragmentation mechanisms. The extended tight binding quantum chemistry package, chosen for its balance between accuracy and computational efficiency, provides molecular geometry optimisation. A Docker image encapsulates the necessary software stack. We demonstrated the workflow for four molecules, highlighting the scalability and efficiency of our solution via runtime performance analysis. This work shows how non-HPC users can make these predictions effortlessly, using advanced computational tools without needing in-depth expertise.
© The Author(s) 2025.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Quantum Chemical Mass Spectral Predictions of Novichok Agents after Experimental Validation and Analysis.ACS Meas Sci Au. 2025 May 23;5(3):378-387. doi: 10.1021/acsmeasuresciau.5c00026. eCollection 2025 Jun 18. ACS Meas Sci Au. 2025. PMID: 40556873 Free PMC article.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
The Lived Experience of Autistic Adults in Employment: A Systematic Search and Synthesis.Autism Adulthood. 2024 Dec 2;6(4):495-509. doi: 10.1089/aut.2022.0114. eCollection 2024 Dec. Autism Adulthood. 2024. PMID: 40018061 Review.
-
Developing a role for patients and the public in the implementation of health and social care research evidence into practice: the PIPER study (Pathways to Implementation for Public Engagement in Research) realist evaluation protocol.Res Involv Engagem. 2025 Jul 14;11(1):80. doi: 10.1186/s40900-025-00728-w. Res Involv Engagem. 2025. PMID: 40660365 Free PMC article. Review.
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
References
-
- Aksenov AA, Da Silva R, Knight R et al. Global chemical analysis of biology by mass spectrometry. Nat. Rev. Chem., 2017; 1: 0054. doi: 10.1038/s41570-017-0054. - DOI
LinkOut - more resources
Full Text Sources