

Shared and disease-specific pathways in frontotemporal dementia and Alzheimer's and Parkinson's diseases
- PMID: 40665052
- PMCID: PMC12404994
- DOI: 10.1038/s41591-025-03833-1
Shared and disease-specific pathways in frontotemporal dementia and Alzheimer's and Parkinson's diseases
Erratum in
-
Author Correction: Shared and disease-specific pathways in frontotemporal dementia and Alzheimer's and Parkinson's diseases.Nat Med. 2025 Oct;31(10):3569. doi: 10.1038/s41591-025-03970-7. Nat Med. 2025. PMID: 40897975 Free PMC article. No abstract available.
Abstract
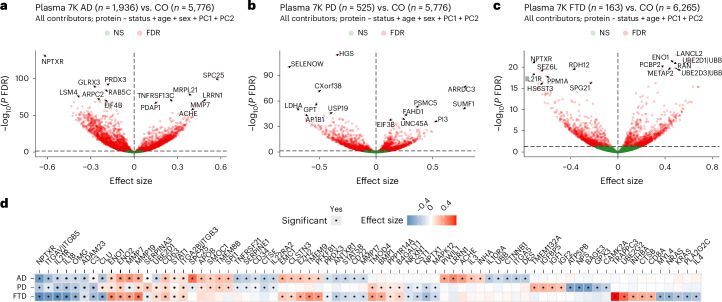
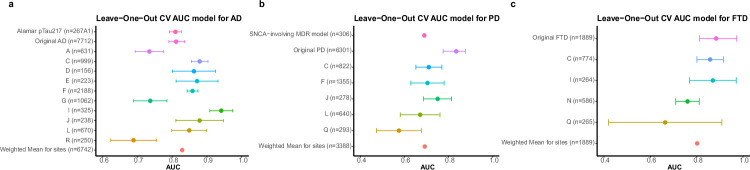
Neurodegenerative diseases (NDs), such as Alzheimer's disease (AD), Parkinson's disease (PD) and frontotemporal dementia (FTD), exhibit distinct yet overlapping pathological mechanisms. Leveraging large-scale plasma proteomics data from the Global Neurodegeneration Proteomics Consortium, we analyzed 10,527 plasma samples (1,936 AD, 525 PD, 163 FTD, 1,638 dementia and 6,265 controls) to identify disease-specific and shared proteins across NDs. We identified 5,187 proteins significantly associated with AD, 3,748 with PD and 2,380 with FTD that revealed both common and divergent proteomic signatures, which were confirmed by multiple analytical approaches and orthogonal validation. PD and FTD showed the highest overlap (r2 = 0.44) and AD and PD the least (r2 = 0.04). Immune system, glycolysis, and matrisome-related pathways were enriched across all NDs, while disease-specific pathways included apoptotic processes in AD, endoplasmic reticulum-phagosome impairment in PD and platelet dysregulation in FTD. Network analysis identified key upstream regulators (RPS27A in AD, IRAK4 in PD and MAPK1 in FTD) potentially driving these proteomic changes. These findings reveal distinct and shared mechanisms across NDs, highlighting potential regulatory proteins and pathways for diagnostic and therapeutic strategies in neurodegeneration.
© 2025. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests: C.C. has received research support from GlaxoSmithKline and EISAI. C.C. is a member of the scientific advisory board of Circular Genomics and owns stocks. C.C. is a member of the scientific advisory board of ADmit. There is an invention disclosure for the prediction models, including protein IDs, alternative proteins and weights, cutoff and algorithms. O.H. is an employee of Eli Lilly and Lund University. M.A., B.E., Y.C., Y.X., K.G., M.L., A.P.B., J.T., D.W., C.Y., G.H., J.W.V., B.M.T., V.K., F.I. and L.W. declare no competing interests.
Figures












References
MeSH terms
Grants and funding
- U01 AG046152/AG/NIA NIH HHS/United States
- 09150171910068/ZonMw (Netherlands Organisation for Health Research and Development)
- P01 AG003991/AG/NIA NIH HHS/United States
- Z01 AG000399/ImNIH/Intramural NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
- R01 AG083740/AG/NIA NIH HHS/United States
- P30 AG072975/AG/NIA NIH HHS/United States
- P30 AG066444/AG/NIA NIH HHS/United States
- U01 AG058922/AG/NIA NIH HHS/United States
- R01 AG044546/AG/NIA NIH HHS/United States
- U01 AG061356/AG/NIA NIH HHS/United States
- RF1 AG053303/AG/NIA NIH HHS/United States
- P01 AG026276/AG/NIA NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- RF1 AG058501/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
