

Molecular characteristics of human adenovirus isolated from the 2024 influenza-like illness outbreaks in Suzhou City, China
- PMID: 40665385
- PMCID: PMC12265240
- DOI: 10.1186/s12985-025-02870-z
Molecular characteristics of human adenovirus isolated from the 2024 influenza-like illness outbreaks in Suzhou City, China
Abstract
Background: Human adenoviruses (HAdV) comprises a genetically diverse group of double-stranded DNA viruses strongly associated with influenza-like illness (ILI) and acute respiratory illnesses (ARI) outbreaks. This study aimed to elucidate the molecular characteristics of HAdV implicated during ILI outbreaks in Suzhou City, Jiangsu Province in 2024.
Methods: Throat swab samples were gathered from settings with ILI outbreaks in Suzhou between March 2024 and January 2025. These samples were analyzed using rapid multi-pathogen detection and real-time PCR. HAdV-positive samples were inoculated into human epidermoid larynx carcinoma cell line (Hep2) for HAdV isolation, followed by whole-genome sequencing (WGS) using the MiSeq platform. Maximum likelihood (ML) phylogenetic trees for the whole genome, hexon, penton base and fiber genes were constructed using IQ-TREE v2.3.6 software. Amino acid variation analysis was performed with BioEdit 7.7.1 software, while recombination detection and visualization were conducted using Recombination Detection Program version 4.101 (RDP4) and SimPlot 3.5.1 software.
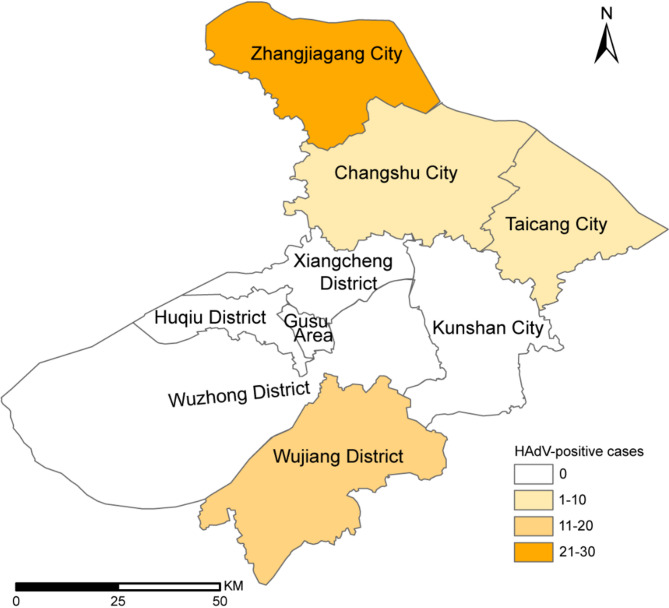
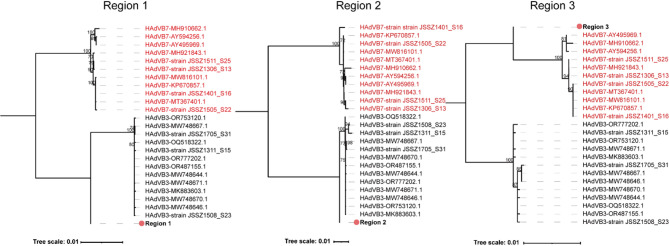
Results: Of 488 outbreak samples collected, 53 (10.86%) tested positive for HAdV, making it the second most prevalent respiratory pathogen. These HAdV-related respiratory outbreaks occurred during the spring-summer and autumn-winter transitions, showing no significant regional differences but a slightly higher positive rate in males than in females (13.83% vs. 7.66%, p = 0.028). Twenty-nine HAdV-positive samples were selected for viral isolation and genome sequencing. Phylogenetic analysis revealed that most isolates clustered with recently circulating HAdVB3 strains, while several clustered with HAdVB7 or previously reported HAdVB3/7 recombinant genotypes. Compared with the HAdVB3 reference sequence (OQ518322.1), multiple nonsynonymous amino acid substitutions were identified in antigenic determinant domains (loop 1 and loop 2) of the hexon protein. Besides, four substitution sites were detected in each of the penton base and fiber proteins. Recombination analysis identified seven strains as HAdVB3/7 recombinants, displaying three distinct recombination patterns. Notably, the strain SZTC20241129 carried HAdVB7-derived insertional fragments spanning partial E2B gene region, 52/55 kDa protein, protein IIIa precursor (pIIIa), and the penton base. In contrast, the other recombinant strains (SZZJ20240513, SZTC20241133, SZZJ20240515, SZZJ20240516, SZCS20241240 and SZCS20241242) possessed an HAdVB7-derived genomic backbone along with multiple HAdVB3-derived insertional fragments, encompassing a range of structural and regulatory genes.
Conclusions: HAdV was one of the leading causative agents of ILI outbreaks in Suzhou in 2024, with HAdVB3 being the predominant genotype (22/29, 75.9%). Strikingly, HAdVB3/7 recombinant strains with P7H7F3 and P7H3F3 genetic constituents were also detected in a subset of cases (7/29, 24.1%). The genetic diversity of circulating HAdV continues to expand, driven by the emergence of novel amino acid substitutions and intertypic recombination events. These findings underscore the importance of sustained molecular surveillance to track viral evolution and inform effective public health strategies aimed at reducing the risk of severe clinical outcomes in vulnerable populations.
Keywords: Genome sequencing; Human adenovirus; Phylogenetic analysis; Recombination; Variation.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: The throat swab samples in this study were collected for infectious disease pathogen testing with the patients’ consent. Genomic sequencing was performed for infectious disease surveillance and public health protection purposes. The interests of the participants involved in this study have been adequately protected, and they were beneficial for the disease diagnosis and treatment. This study was approved by Suzhou Center for Disease Control and Prevention (CDC) ethics committee. All experiments were in line with relevant rules and regulations. Consent for publication: All authors agree to publish the research data. Competing interests: The authors declare no competing interests.
Figures
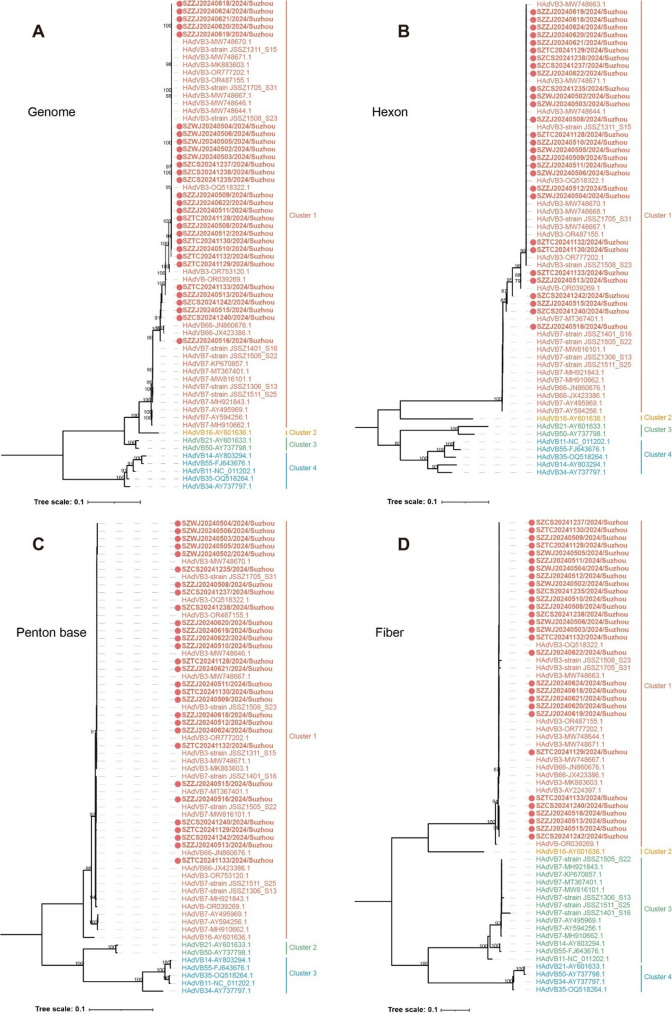
 ). Numbers above branches indicate bootstrap values, with those greater than 75% were shown
). Numbers above branches indicate bootstrap values, with those greater than 75% were shown
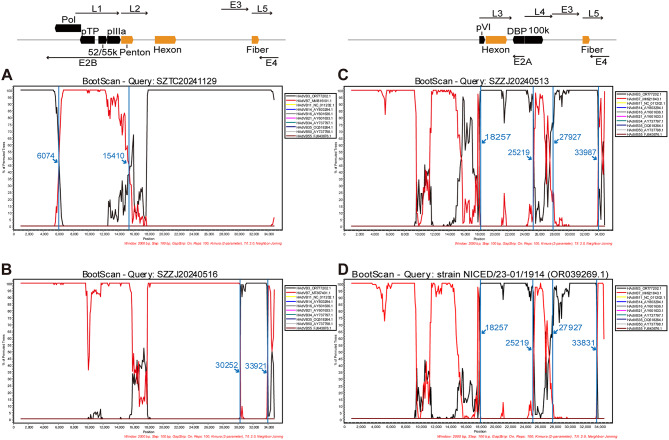
 ). Numbers above branches indicate bootstrap values, with those greater than 70% were shown
). Numbers above branches indicate bootstrap values, with those greater than 70% were shownReferences
Publication types
MeSH terms
Associated data
- Actions
LinkOut - more resources
Full Text Sources
