

Expectation Value-pCCD-Based Methods for Single-Electron Properties
- PMID: 40665493
- PMCID: PMC12302219
- DOI: 10.1021/acs.jpca.5c03859
Expectation Value-pCCD-Based Methods for Single-Electron Properties
Abstract
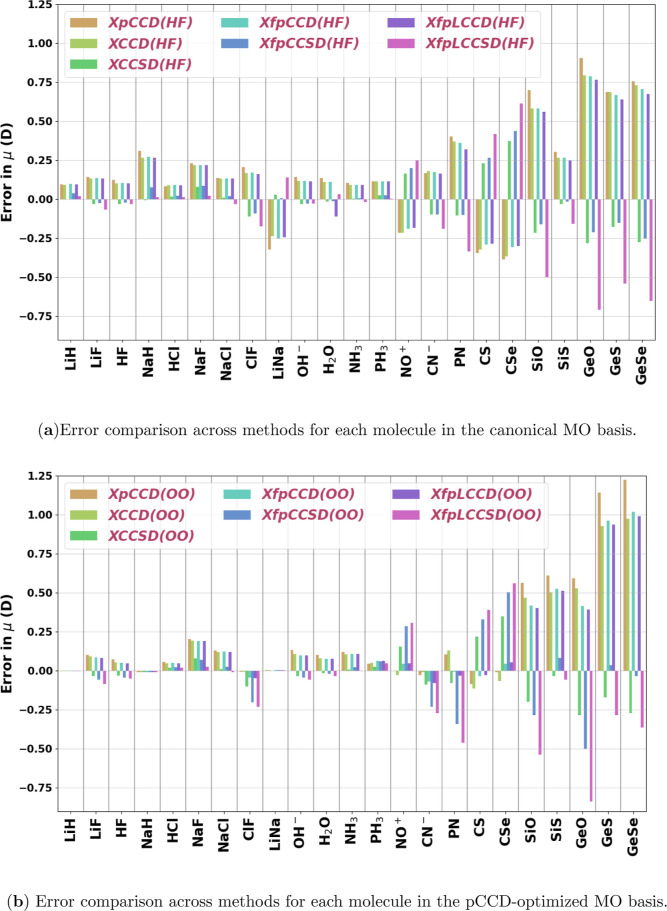
Expectation-value-coupled cluster theory (XCC) offers a simple avenue for molecular property evaluation. However, its potential has not been fully explored for the new computationally inexpensive CC models, such as pair-coupled cluster doubles (pCCD) and post-pCCD extensions. To that end, we implemented and explored one-electron reduced density matrices in the explicitly connected commutator expansion of the expectation value framework [J. Chem. Phys. 2006, 125, 184109] using pCCD, frozen pair Coupled Cluster (fpCC), and frozen pair linearized Coupled Cluster (fpLCC) variants. The expectation-value-based density matrices are calculated directly using the cluster amplitudes and are computationally cheaper than the corresponding response CC densities, as we bypass solving the computationally expensive Λ-equations. The performance of this approach, when combined with the pCCD-based methods, is assessed against the dipole and quadrupole moments of molecules of a varying chemical nature. We benchmarked our results against the response of CCSD(T) using Hartree-Fock canonical orbitals and variationally optimized pCCD orbitals. Our study highlights that localized pCCD orbitals are a good choice for computing one-electron properties of organic molecules.
Figures









References
-
- Čižek J.. On the Correlation Problem in Atomic and Molecular Systems. Calculation of Wavefunction Components in Ursell-Type Expansion Using Quantum-Field Theoretical Methods. J. Chem. Phys. 1966;45:4256–4266. doi: 10.1063/1.1727484. - DOI
-
- Čižek J.. On the use of the cluster expansion and the technique of diagrams in calculations of correlation effects in atoms and molecules. Adv. Chem. Phys. 1969;14:35–89. doi: 10.1002/9780470143599.ch2. - DOI
-
- Čižek J., Paldus J.. Correlation problems in atomic and molecular systems III. Rederivation of the coupled-pair many-electron theory using the traditional quantum chemical methods. Int. J. Quantum Chem. 1971;5:359–379. doi: 10.1002/qua.560050402. - DOI
-
- Bartlett R. J.. Many-Body Perturbation Theory and Coupled Cluster Theory for Electron Correlation in Molecules. Annu. Rev. Phys. Chem. 1981;32:359–401. doi: 10.1146/annurev.pc.32.100181.002043. - DOI
-
- Bartlett R. J., Musiał M.. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007;79:291–350. doi: 10.1103/RevModPhys.79.291. - DOI
LinkOut - more resources
Full Text Sources
