

Interpretable deep learning framework for understanding molecular changes in human brains with Alzheimer's disease: implications for microglia activation and sex differences
- PMID: 40670382
- PMCID: PMC12267417
- DOI: 10.1038/s41514-025-00258-5
Interpretable deep learning framework for understanding molecular changes in human brains with Alzheimer's disease: implications for microglia activation and sex differences
Abstract
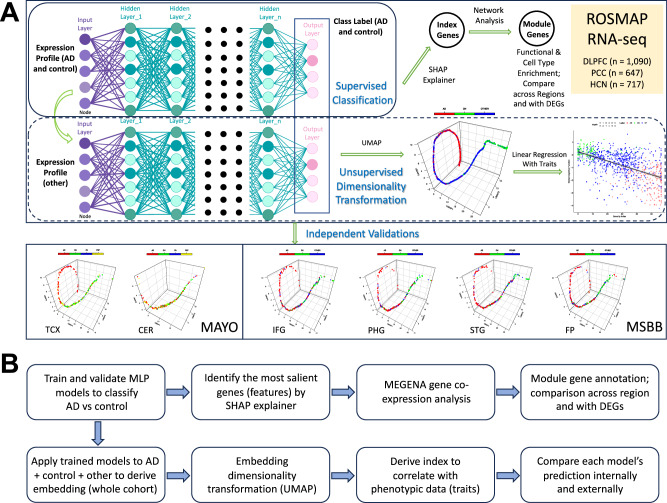
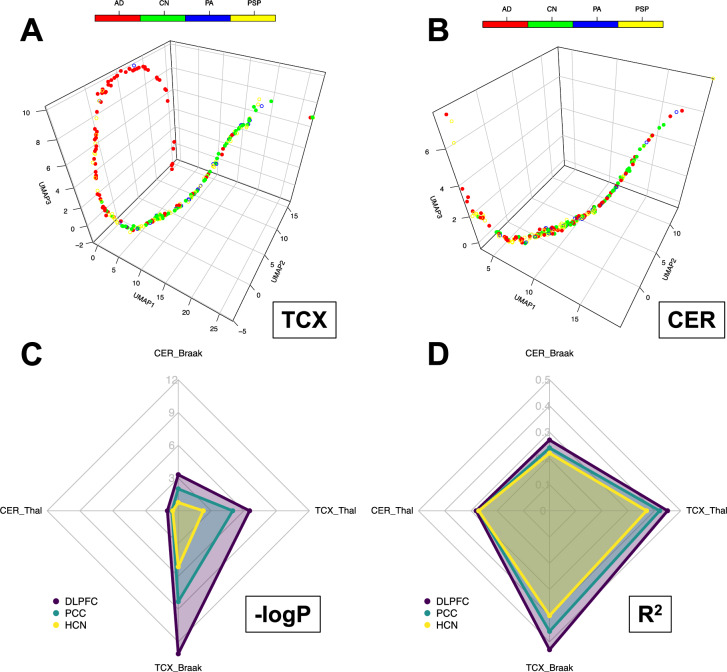
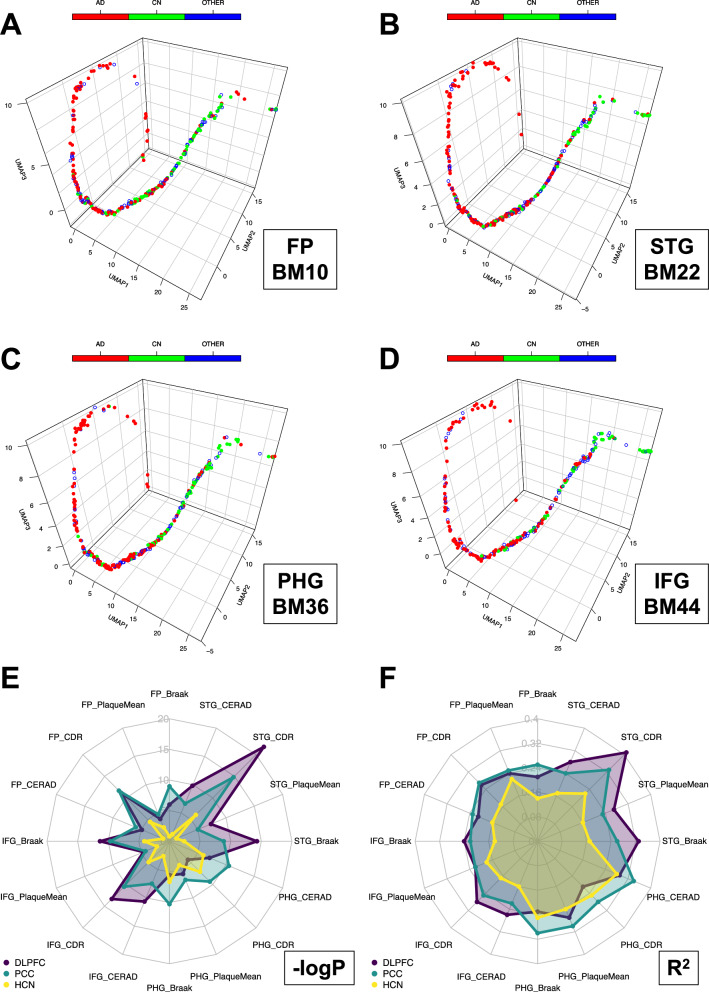
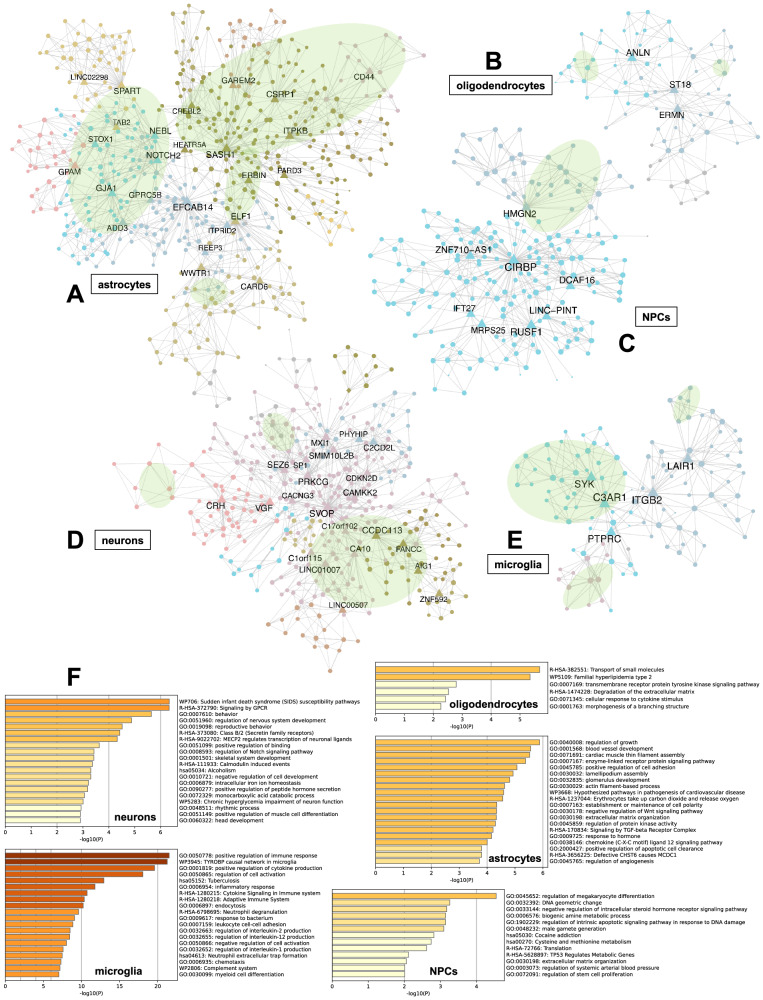
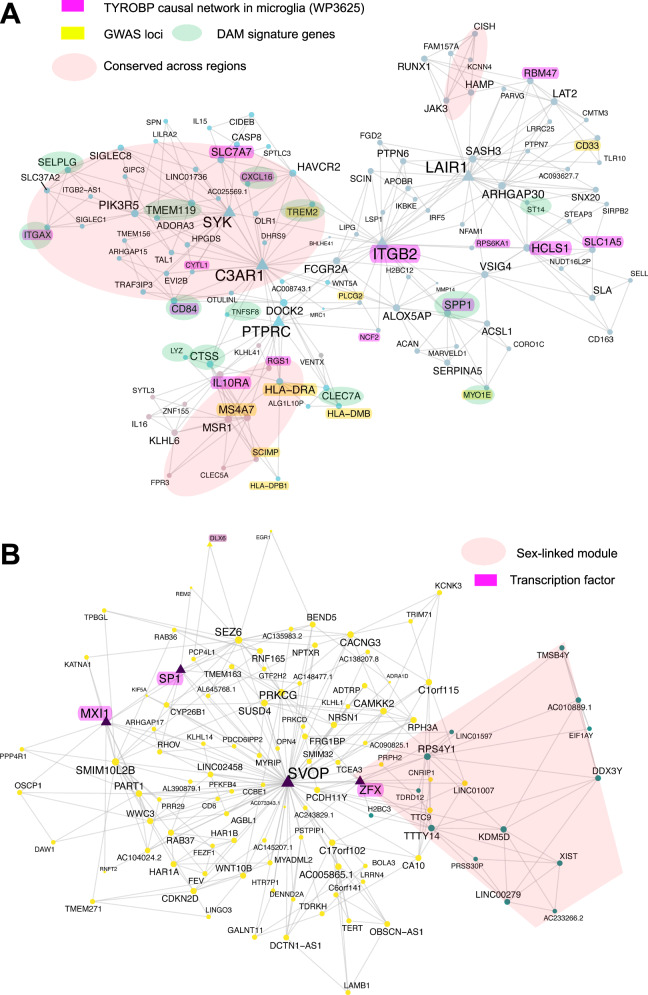
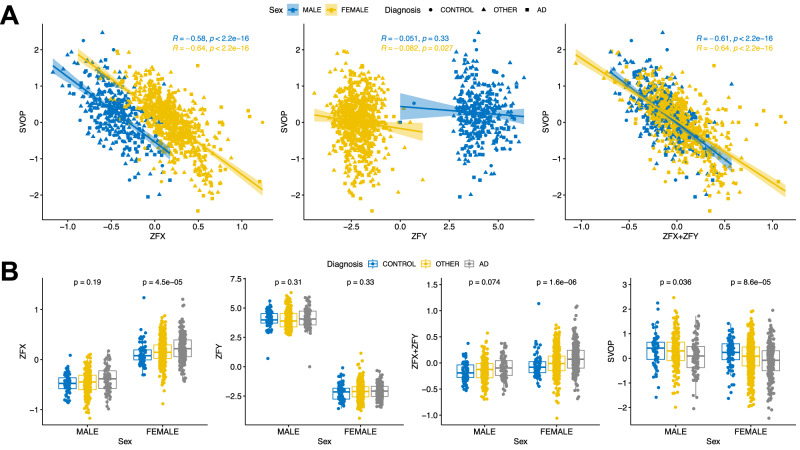
The utilization of artificial intelligence in studying the dysregulation of gene expression in Alzheimer's disease (AD) affected brain tissues remains underexplored, particularly in delineating common and specific transcriptomic signatures across different brain regions implicated in AD-related cellular and molecular processes, which could help illuminate novel disease biology for biomarker and target discovery. Herein we developed a deep learning framework, which consisted of multi-layer perceptron (MLP) models to classify neuropathologically confirmed AD versus controls, using bulk tissue RNA-seq data from the RNAseq Harmonization Study of the Accelerating Medicines Project for Alzheimer's Disease (AMP-AD) consortium. The models were trained based on data from three distinct brain regions, including dorsolateral prefrontal cortex (DLPFC), posterior cingulate cortex (PCC), and head of the caudate nucleus (HCN), obtained from the Religious Orders Study/Memory and Aging Project (ROSMAP). Subsequently, we inferred a disease progression trajectory for each brain region by applying unsupervised dimensionality transformation to the distribution of the subjects' expression profiles. To interpret the MLP models, we employed an interpretable method for deep neural network models, obtaining SHapley Additive exPlanations (SHAP) values and identified the most significantly AD-implicated genes for gene co-expression network analysis. Our models demonstrated robust performance in classification and prediction across two other external datasets from the Mayo RNA-seq (MAYO) cohort and the Mount Sinai Brain Bank (MSBB) cohort of AMP-AD. By interpreting the models both mechanistically and biologically, our study elucidated subtle molecular alterations in various brain regions, uncovering shared transcriptomic signatures activated in microglia and sex-specific modules in neurons relevant to AD. Notably, we identified, for the first time, a sex-linked transcription factor pair (ZFX/ZFY) associated with more pronounced neuronal loss in AD females, shedding light on a novel mechanism for sex dimorphism in AD. This study lays the groundwork for leveraging artificial intelligence methodologies to investigate AD at the molecular level, which is not readily achievable from conventional analysis approaches such as differential gene expression (DGE) analysis. The transcription factor implicated in sex difference also underpins a new molecular mechanistic basis of women's greater neurodegeneration in AD warranting further study.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures

Similar articles
-
Predicting cognitive decline: Deep-learning reveals subtle brain changes in pre-MCI stage.J Prev Alzheimers Dis. 2025 May;12(5):100079. doi: 10.1016/j.tjpad.2025.100079. Epub 2025 Feb 6. J Prev Alzheimers Dis. 2025. PMID: 39920001 Free PMC article.
-
From aging to Alzheimer's disease: concordant brain DNA methylation changes in late life.medRxiv [Preprint]. 2025 Jun 18:2025.06.17.25329345. doi: 10.1101/2025.06.17.25329345. medRxiv. 2025. PMID: 40585142 Free PMC article. Preprint.
-
Comprehensive characterization of the RNA editing landscape in the human aging brains with Alzheimer's disease.Alzheimers Dement. 2025 Jul;21(7):e70452. doi: 10.1002/alz.70452. Alzheimers Dement. 2025. PMID: 40631452 Free PMC article.
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
References
-
- Santorsola, M. & Lescai, F. The promise of explainable deep learning for omics data analysis: Adding new discovery tools to AI. N. Biotechnol.77, 1–11 (2023). - PubMed
Grants and funding
- P30AG072980/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- U01AG061835/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- U01AG061835/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- RF1AG073424/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- P30AG072980/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
LinkOut - more resources
Full Text Sources
