

Ongoing genome doubling shapes evolvability and immunity in ovarian cancer
- PMID: 40670783
- PMCID: PMC12390843
- DOI: 10.1038/s41586-025-09240-3
Ongoing genome doubling shapes evolvability and immunity in ovarian cancer
Abstract
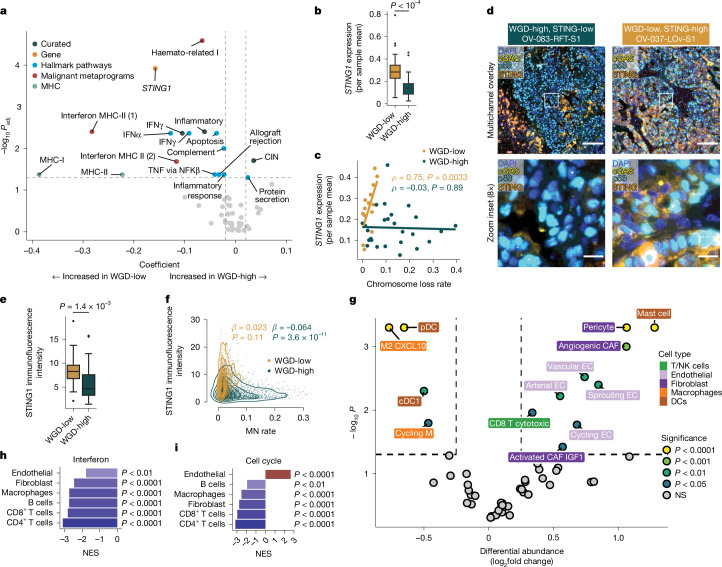
Whole-genome doubling (WGD) is a common feature of human cancers and is linked to tumour progression, drug resistance, and metastasis1-6. Here we examine the impact of WGD on somatic evolution and immune evasion at single-cell resolution in patient tumours. Using single-cell whole-genome sequencing, we analysed 70 high-grade serous ovarian cancer samples from 41 patients (30,260 tumour genomes) and observed near-ubiquitous evidence that WGD is an ongoing mutational process. WGD was associated with increased cell-cell diversity and higher rates of chromosomal missegregation and consequent micronucleation. We developed a mutation-based WGD timing method called doubleTime to delineate specific modes by which WGD can drive tumour evolution, including early fixation followed by considerable diversification, multiple parallel WGD events on a pre-existing background of copy-number diversity, and evolutionarily late WGD in small clones and individual cells. Furthermore, using matched single-cell RNA sequencing and high-resolution immunofluorescence microscopy, we found that inflammatory signalling and cGAS-STING pathway activation result from ongoing chromosomal instability, but this is restricted to predominantly diploid tumours (WGD-low). By contrast, predominantly WGD tumours (WGD-high), despite increased missegregation, exhibited cell-cycle dysregulation, STING1 repression, and immunosuppressive phenotypic states. Together, these findings establish WGD as an ongoing mutational process that promotes evolvability and dysregulated immunity in high-grade serous ovarian cancer.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: B.W. reports grant funding by Repare Therapeutics paid to the institution, outside the scope of this paper, and employment of a direct family member at AstraZeneca. C.A. reports grants from Clovis, Genentech, AbbVie and AstraZeneca, and personal fees from Tesaro, Eisai/Merck, Mersana Therapeutics, Roche/Genentech, Abbvie, AstraZeneca/Merck and Repare Therapeutics, outside the scope of this paper. C.A. also reports clinical-trial funding to the institution from Abbvie, AstraZeneca and Genentech/Roche; participation on a data safety monitoring board or advisory board at AstraZeneca and Merck; unpaid membership of the GOG Foundation board of directors and the NRG Oncology board of directors. C.F.F. reports research funding to the institution from Merck, AstraZeneca, Genentech/Roche, Bristol Myers Squibb and Daiichi; uncompensated membership of a scientific advisory board for Merck and Genentech; and is a consultant for OncLive, Aptitude Health, Bristol Myers Squibb and Seagen, all outside the scope of this paper. D.S.C. reports membership of the medical advisory board of Verthermia Acquio and Biom’up, is a paid speaker for AstraZeneca and holds stock of Doximity, Moderna and BioNTech. D.Z. reports institutional grants from Merck, Genentech, AstraZeneca, Plexxikon and Synthekine, and personal fees from AstraZeneca, Xencor, Memgen, Takeda, Astellas, Immunos, Tessa Therapeutics, Miltenyi and Calidi Biotherapeutics. D.Z. holds a patent on the use of oncolytic Newcastle disease virus for cancer therapy. N.R.A.-R. reports grants to the institution from Stryker/Novadaq and GRAIL, outside the scope of this paper. R.N.G. reports funding from GSK, Novartis, Mateon Therapeutics, Corcept, Regeneron, Clovis, Context Therapeutics, EMD Serono, MCM Education, OncLive, Aptitude Health and Prime Oncology, outside the scope of this paper. S.F.B. owns equity in, receives compensation from and serves as a consultant and on the scientific advisory board and board of directors of Volastra Therapeutics. He also serves on the scientific advisory board of Meliora Therapeutics. S.P.S. reports research funding from AstraZeneca and Bristol Myers Squibb, outside the scope of this paper. Y.L.L. reports research funding from AstraZeneca, GSK/Tesaro, Artios Pharma and Tesaro Therapeutics, outside the scope of this paper. Y.L. reports serving as a consultant for Calyx Clinical Trial Solutions, outside the scope of this paper. All the remaining authors declare no competing interests.
Figures













References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
