

Mitochondria from huntington´s disease striatal astrocytes are hypermetabolic and compromise neuronal branching
- PMID: 40671084
- PMCID: PMC12265250
- DOI: 10.1186/s12964-025-02341-6
Mitochondria from huntington´s disease striatal astrocytes are hypermetabolic and compromise neuronal branching
Abstract
Background: Deficits in mitochondrial bioenergetics and dynamics are strongly implicated in the selective vulnerability of striatal neurons in Huntington´s disease. Beyond these neuron-intrinsic factor, increasing evidence suggest that non-neuronal mechanisms, particularly astrocytic dysfunction involving disrupted homeostasis and metabolic support also contribute to disease progression. These findings underscore the critical role of metabolic crosstalk between neurons and astrocytes in maintaining striatal integrity. However, it remains unclear whether this impaired communication affects the transfer of mitochondria from astrocytes to striatal neurons, a potential metabolic support mechanism that may be compromised in Huntington´s Disease.
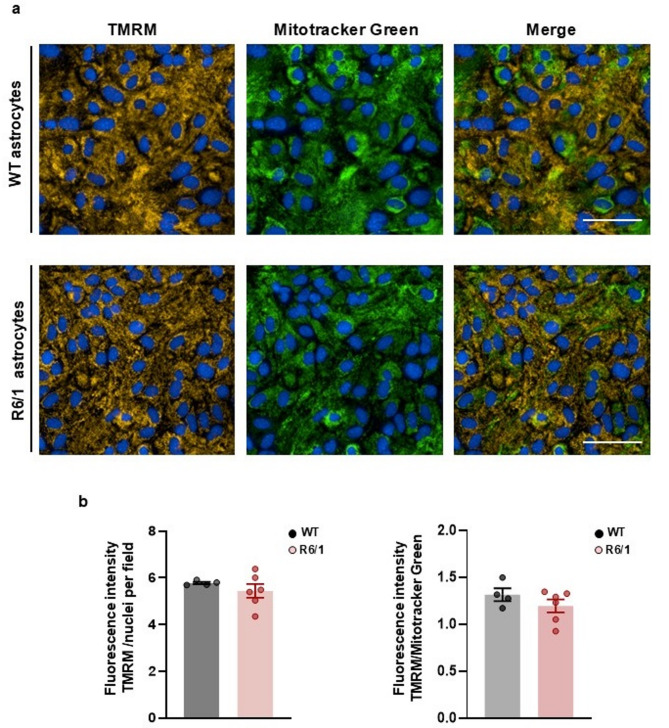
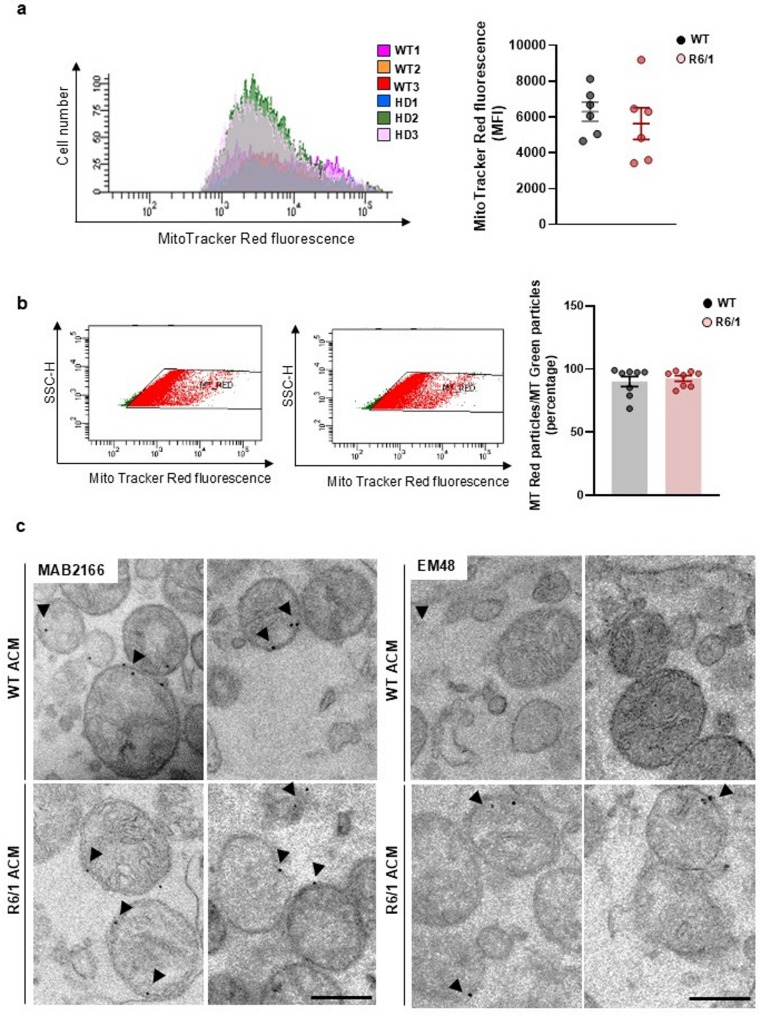
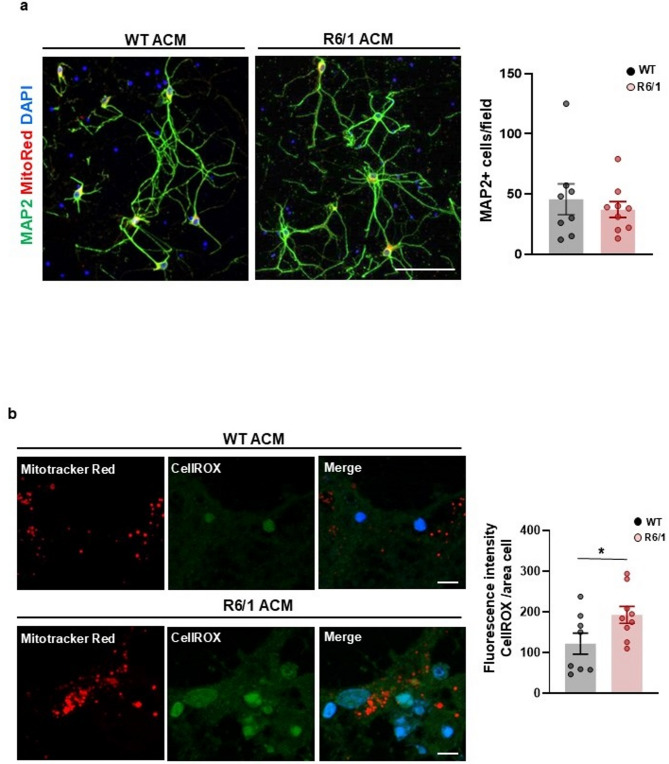
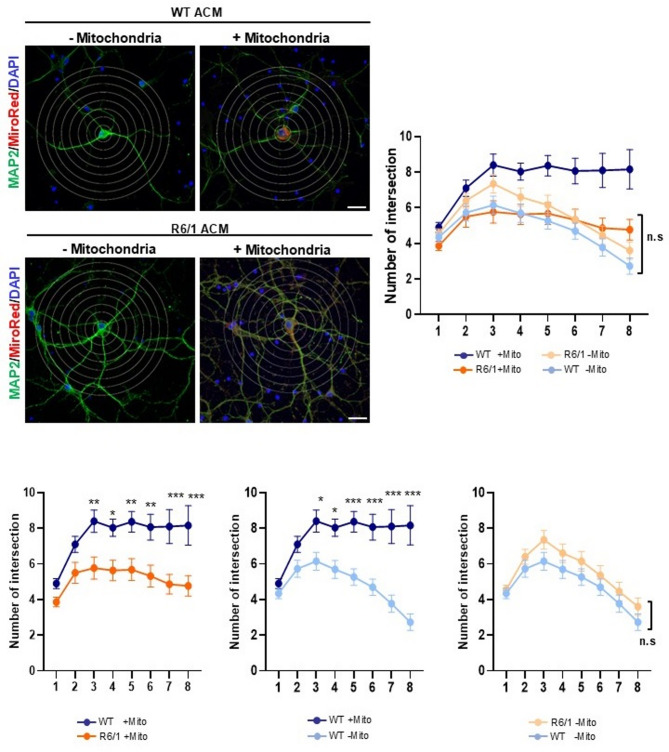
Methods: Primary striatal astrocytes were obtained from wild-type and R6/1 mice to investigate mitochondrial dynamics. Expression levels of key mitochondrial fusion and fission proteins were quantified by Western blotting and RT-PCR. Mitochondria morphology, oxidative stress and membrane potential were assessed using confocal microscopy following staining with mitochondria-specific dyes. Mitochondrial respiration was measured using the Oxygraph-2k respirometer system (Oroboros Instruments). Transmitophagy was evaluated by confocal imaging after labeling astrocytic mitochondria with Mitotracker dyes. To assess the functional impact of mitochondrial transfer on neurons, Sholl analysis, neuronal death and oxidative stress levels were quantified using specific fluorogenic probes.
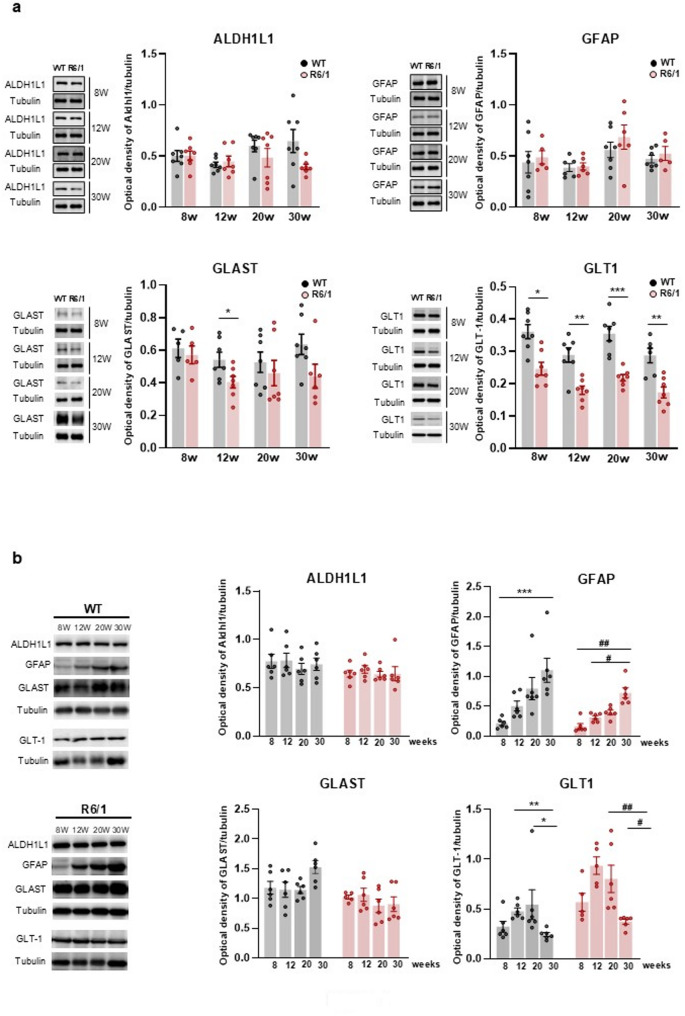
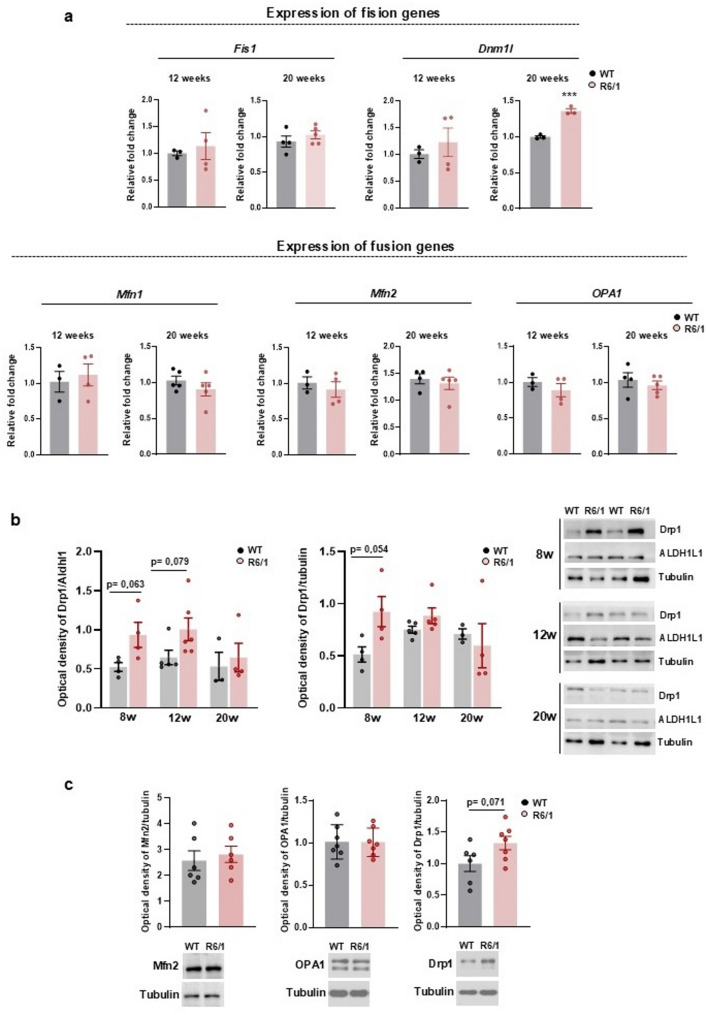
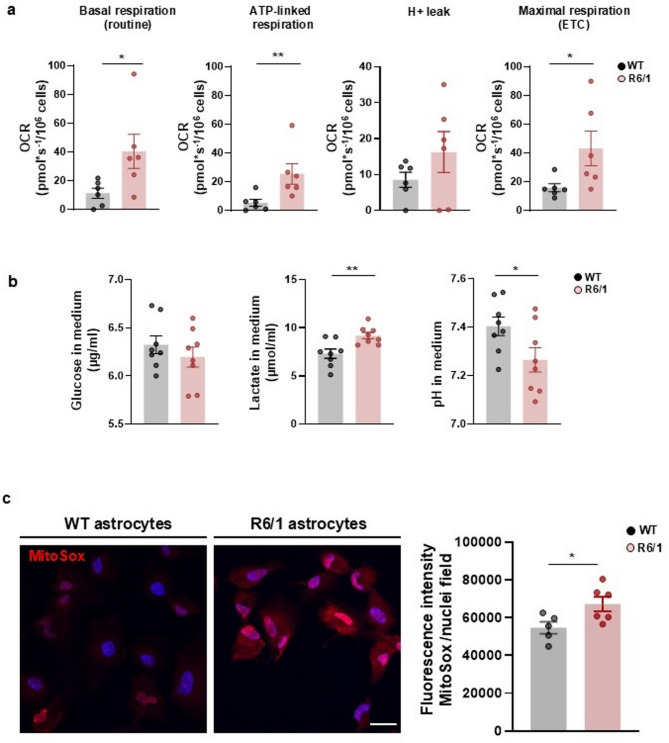
Results: Striatal astrocytes from HD mice exhibited a significant increase in mitochondrial fission, and mitochondrial oxidative stress, mirroring alterations previously reported in striatal neurons. Analysis of mitochondrial oxygen consumption rate (OCR) revealed elevated respiration activity and enhanced ATP-linked respiration, indicative of a hypermetabolic state. Concurrently, increased lactate production suggested a shift toward dysregulated astrocytic energy metabolism. These mitochondrial alterations were functionally detrimental: astrocytic mitochondria derived from HD mice when taken up by striatal neurons via transmitophagy, led to reduced neuronal branching and disrupted oxidative homeostasis.
Conclusions: Our findings demonstrate that striatal astrocytes from HD mice exhibit a hypermetabolic phenotype, characterized by increased mitochondrial respiration, disrupted mitochondrial dynamics, and elevated mitochondrial oxidative stress. Importantly, we identify a novel mechanism of astrocyte-neuron interaction involving the transfer of dysfunctional mitochondria from astrocytes to neurons. The uptake of these compromised mitochondria by striatal neurons results in reduced neuronal branching and increased reactive oxygen species (ROS) production. Collectively, these results highlight the pathological relevance of impaired astrocyte-to-neuron mitochondrial transfer and emphasize the contributory role of astrocytic dysfunction in Huntington´s disease progression.
Keywords: Astrocytes; Huntingtin; Mitochondria transfer; Neuroglial communication; R6/1 mice; Striatum.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Experimental procedures with mice were approved by the Local Ethical Committee of the University of Barcelona (CEEA 369/19 and 439/22) and the Generalitat de Catalunya (DAAM 369/19), following European (86/609/EU) and Spanish (RD 1201/2005) regulations for the care and use of laboratory animals. Consent for publication: Not applicable. Competing interests: The authors declare no competing interests.
Figures

References
-
- MacDonald M. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntington’s disease chromosomes. Cell. 1993;72:971–83. - PubMed
-
- Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. - PubMed
-
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. Neuropathological classification of huntingtonʼs disease. J Neuropathol Exp Neurol. 1985;44:559–77. - PubMed
-
- Vonsattel JPG, Keller C, Cortes Ramirez EP. Huntington’s disease– neuropathology. 2011. pp. 83–100. - PubMed
-
- Mehrabi NF, Waldvogel HJ, Tippett LJ, Hogg VM, Synek BJ, Faull RLM. Symptom heterogeneity in huntington’s disease correlates with neuronal degeneration in the cerebral cortex. Neurobiol Dis. 2016;96:67–74. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
