

This is a preprint.
Autophagy activators normalize aberrant Tau proteostasis and rescue synapses in human familial Alzheimer's disease iPSC-derived cortical organoids
- PMID: 40672178
- PMCID: PMC12265565
- DOI: 10.1101/2025.06.25.661453
Autophagy activators normalize aberrant Tau proteostasis and rescue synapses in human familial Alzheimer's disease iPSC-derived cortical organoids
Abstract
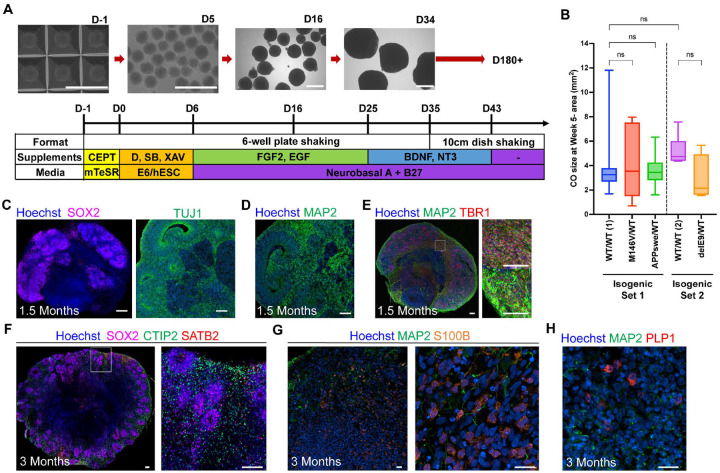
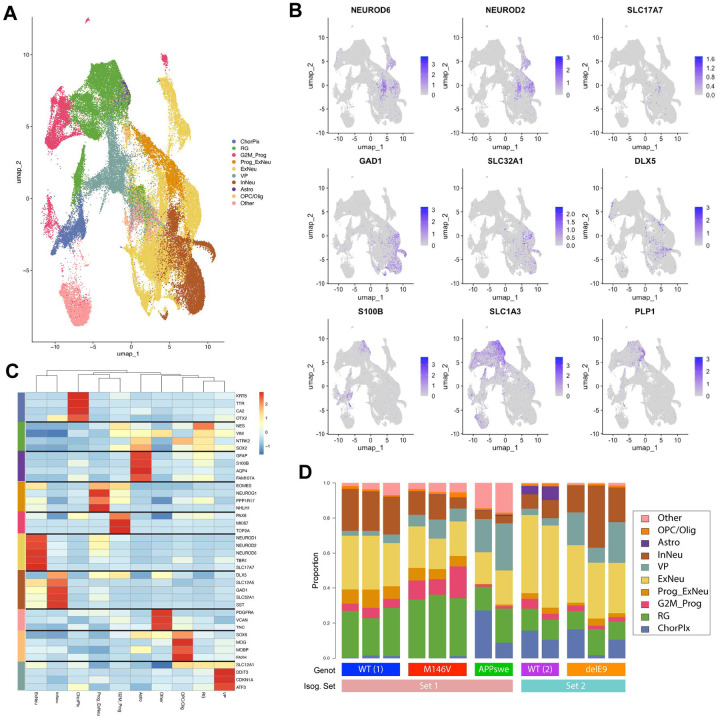
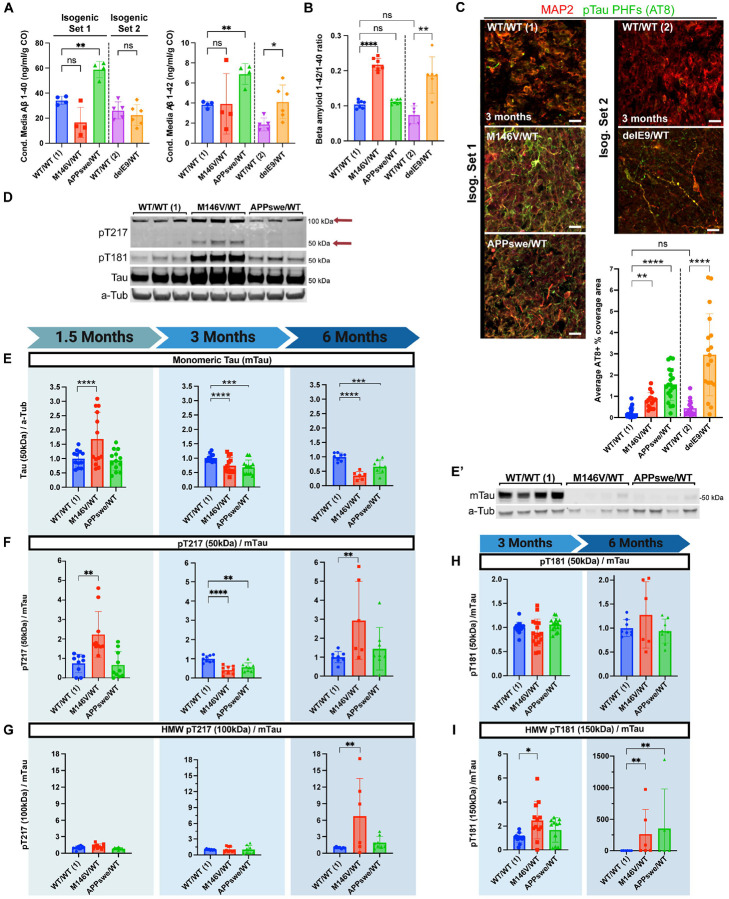
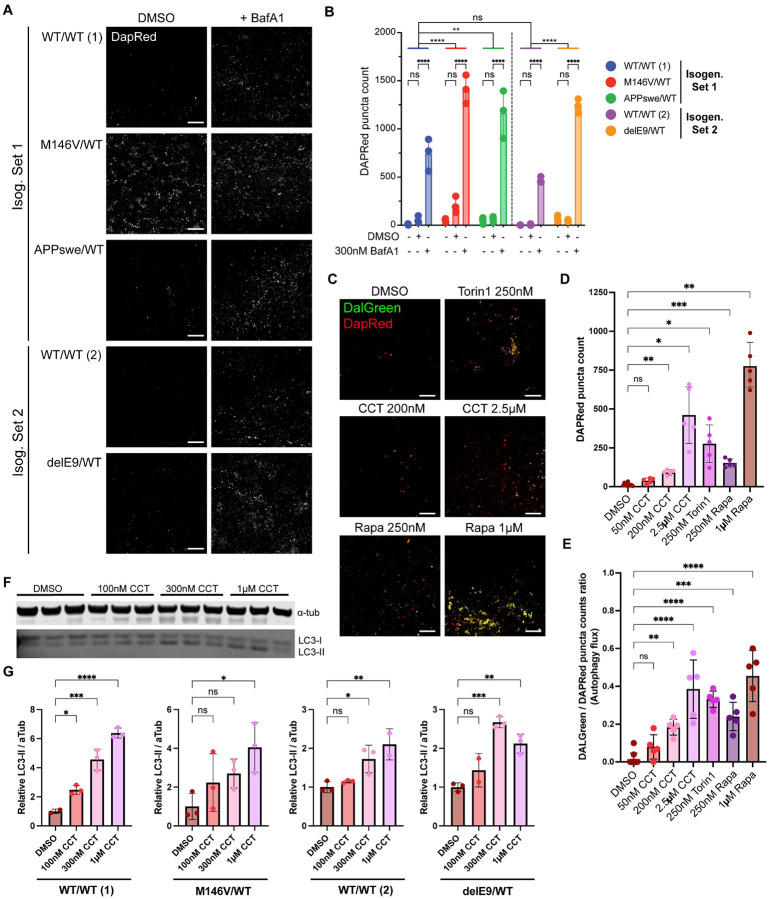
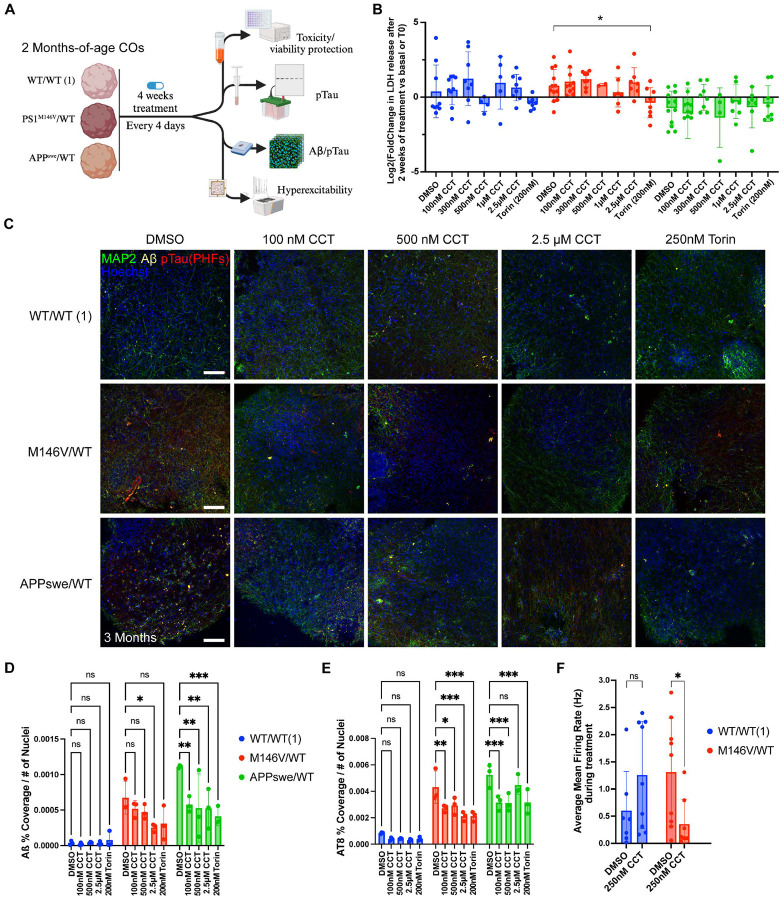
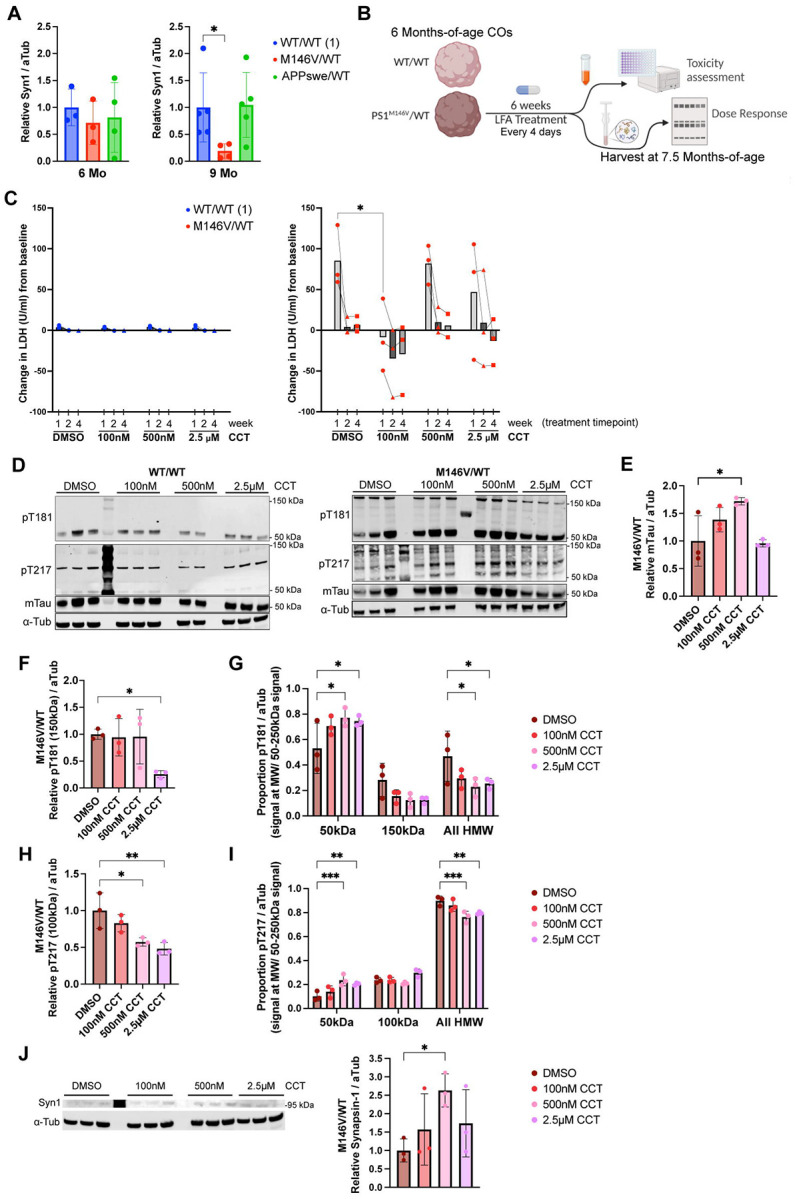
Alzheimer's disease (AD) is the most common form of dementia worldwide. Despite extensive progress, the cellular and molecular mechanisms of AD remain incompletely understood, partially due to inadequate disease models. To illuminate the earliest changes in hereditary (familial) Alzheimer's disease, we developed an isogenic AD cerebrocortical organoid (CO) model. Our refined methodology produces COs containing excitatory and inhibitory neurons alongside glial cells, utilizing established isogenic wild-type and diseased human induced pluripotent stem cells (hiPSCs) carrying heterozygous familial AD mutations, namely PSEN1ΔE9/WT, PSEN1M146V/WT, or APPswe/WT. Our CO model reveals time-progressive accumulation of amyloid beta (Aβ) species, loss of monomeric Tau, and accumulation of aggregated high-molecular-weight (HMW) phospho(p)-Tau species. This is accompanied by neuronal hyperexcitability, as observed in early human AD cases on electroencephalography (EEG), and synapse loss. Single-cell RNA-sequencing analyses reveal significant differences in molecular abnormalities in excitatory vs. inhibitory neurons, helping explain AD clinical phenotypes. Finally, we show that chronic dosing with autophagy activators, including a novel CNS-penetrant mTOR inhibitor-independent drug candidate, normalizes pathologic accumulation of Aβ and HMW p-Tau, normalizes hyperexcitability, and rescues synaptic loss in COs. Collectively, our results demonstrate these COs are a useful human AD model suitable for assessing early features of familial AD etiology and for testing drug candidates that ameliorate or prevent molecular AD phenotypes.
Keywords: Alzheimer’s disease; Amyloid Beta Precursor Protein; Autophagy; Cortical organoids; Presenilin 1; Tau; chronic treatment; human model; iPSC-derived; oligomers; pTau.
Conflict of interest statement
DECLARATION OF INTERESTS J.W.K. discloses that he receives royalties for Tafamidis sales as an inventor and has received additional payments from Pfizer. J.W.K is a founder and major shareholder of Protego, which is developing immunoglobulin light chain kinetic stabilizers and other stabilizers for misfolding diseases; he serves on its Board of Directors and Scientific Advisory Board and acts as a consultant. He serves as a consultant for the Dominantly Inherited Alzheimer Network Trial Unit in reviewing drug candidates. S.A.L discloses that he is an inventor on worldwide patents for the use of memantine and NitroSynapsin (aka NitroMemantine, YQW-036, or EM-036) for neurodegenerative and neurodevelopmental disorders. Per Harvard University guidelines, S.A.L. participates in a royalty-sharing agreement with his former institution Boston Children’s Hospital/Harvard Medical School, which licensed the drug memantine (Namenda®) to Forest Laboratories, Inc./Actavis/Allergan/AbbVie. S.A.L. was scientific founder of Adamas Pharmaceuticals, Inc. (now owned by Supernus Pharmaceuticals, Inc.), which developed or comarketed FDA-approved forms of memantine- or amantadine-containing drugs (NamendaXR®, Namzaric®, and GoCovri®). NitroSynapsin is licensed to the biotechnology company EuMentis Therapeutics, Inc., for which SAL is scientific founder and chair of the Scientific Advisory Board (SAB). SAL is also a member of the SAB of Point 6 Bio. Ltd., and has recently served as a consultant to Circumvent Pharmaceuticals, Inc. Further, SAL discloses that he is a named inventor on patent(s) filed by his current institution, The Scripps Research Institute, for novel MEF2 and NRF2 transcriptional activators in the treatment of systemic and nervous system diseases via neuroprotective, anti-inflammatory, and antioxidant actions
Figures
References
-
- Cullen N.C., Novak P., Tosun D., Kovacech B., Hanes J., Kontsekova E., Fresser M., Ropele S., Feldman H.H., and Schmidt R. (2024). Efficacy assessment of an active tau immunotherapy in Alzheimer’s disease patients with amyloid and tau pathology: A post hoc analysis of the “ADAMANT” randomised, placebo-controlled, double-blind, multi-centre, phase 2 clinical trial. EBioMedicine 99. - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous