

This is a preprint.
Context-dependent regulatory variants in Alzheimer's disease
- PMID: 40672290
- PMCID: PMC12265656
- DOI: 10.1101/2025.07.11.659973
Context-dependent regulatory variants in Alzheimer's disease
Abstract
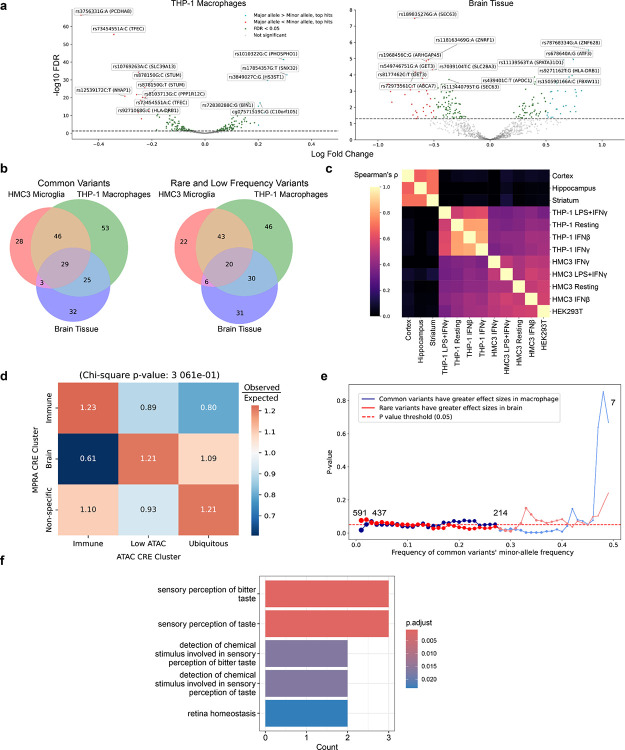
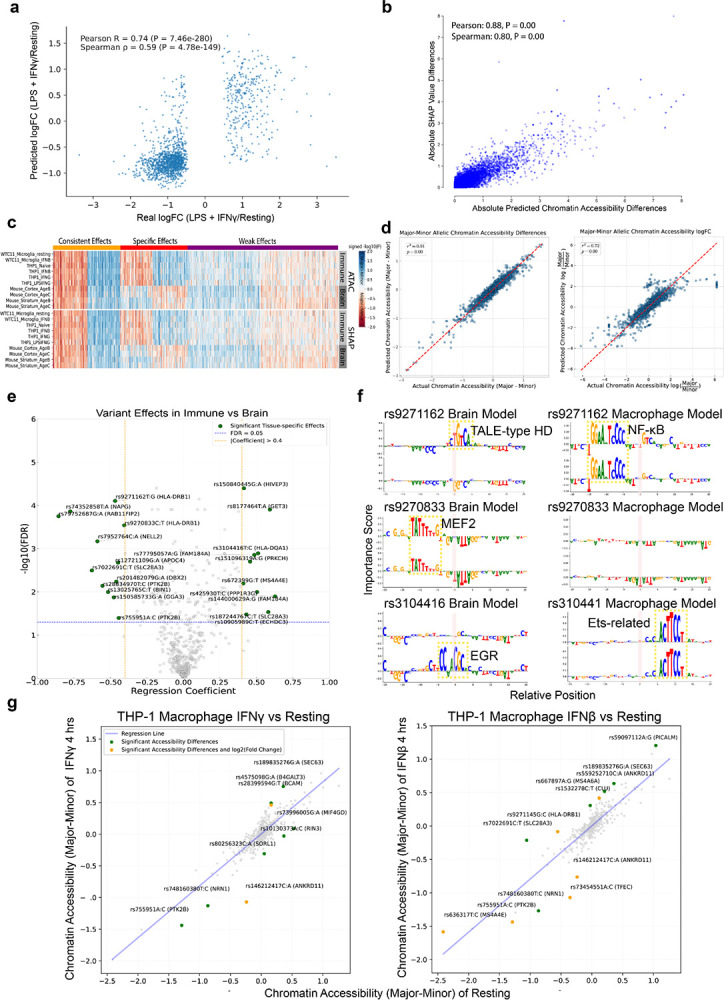
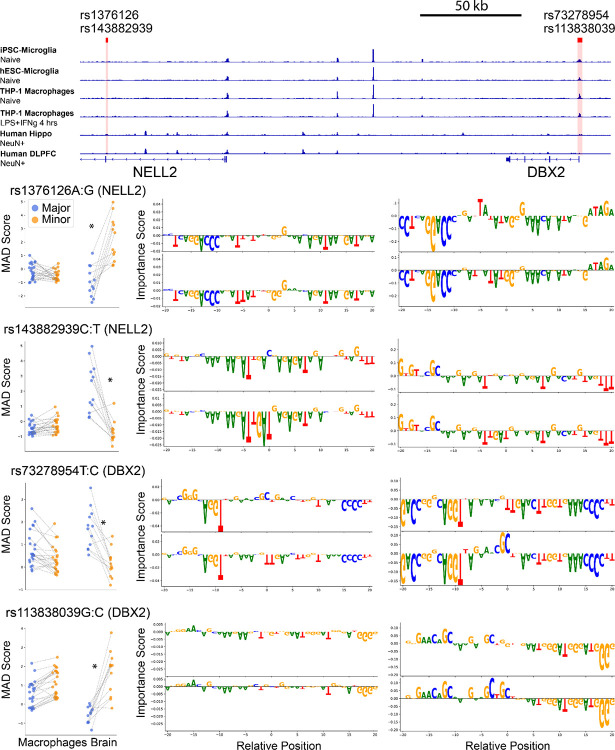
Noncoding genetic variants underlie many complex diseases, yet identifying and interpreting their functional impacts remains challenging. Late-onset Alzheimer's disease (LOAD), a polygenic neurodegenerative disorder, exemplifies this challenge. The disease is strongly associated with noncoding variation, including common variants enriched in microglial enhancers and rare variants that are hypothesized to influence neurodevelopment and synaptic plasticity. These variants often perturb regulatory sequences by disrupting transcription factor (TF) motifs or altering local TF interactions, thereby reshaping gene expression and chromatin accessibility. However, assessing their impact is complicated by the context-dependent functions of regulatory sequences, underscoring the need to systematically examine variant effects across diverse tissues, cell types, and cellular states. Here, we combined in vitro and in vivo massively parallel reporter assays (MPRAs) with interpretable machine-learning models to systematically characterize common and rare variants across myeloid and neural contexts. Parallel profiling of variants in four immune states in vitro and three mouse brain regions in vivo revealed that individual variants can differentially and even oppositely modulate regulatory function depending on cell-type and cell-state contexts. Common variants associated with LOAD tended to exert stronger effects in immune contexts, whereas rare variants showed more pronounced impacts in brain contexts. Interpretable sequence-to-function deep-learning models elucidated how genetic variation leads to cell-type-specific differences in regulatory activity, pinpointing both direct transcription-factor motif disruptions and subtler tuning of motif context. To probe the broader functional consequences of a locus prioritized by our reporter assays and models, we used CRISPR interference to silence an enhancer within the SEC63-OSTM1 locus that harbors four functional rare variants, revealing its gatekeeper role in inflammation and amyloidogenesis. These findings underscore the context-dependent nature of noncoding variant effects in LOAD and provide a generalizable framework for the mechanistic interpretation of risk alleles in complex diseases.
Conflict of interest statement
Competing Interests None.
Figures











References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous