

Long-read sequencing to detect full-length protein-protein interactions
- PMID: 40676061
- PMCID: PMC12271486
- DOI: 10.1038/s41598-025-08549-3
Long-read sequencing to detect full-length protein-protein interactions
Abstract
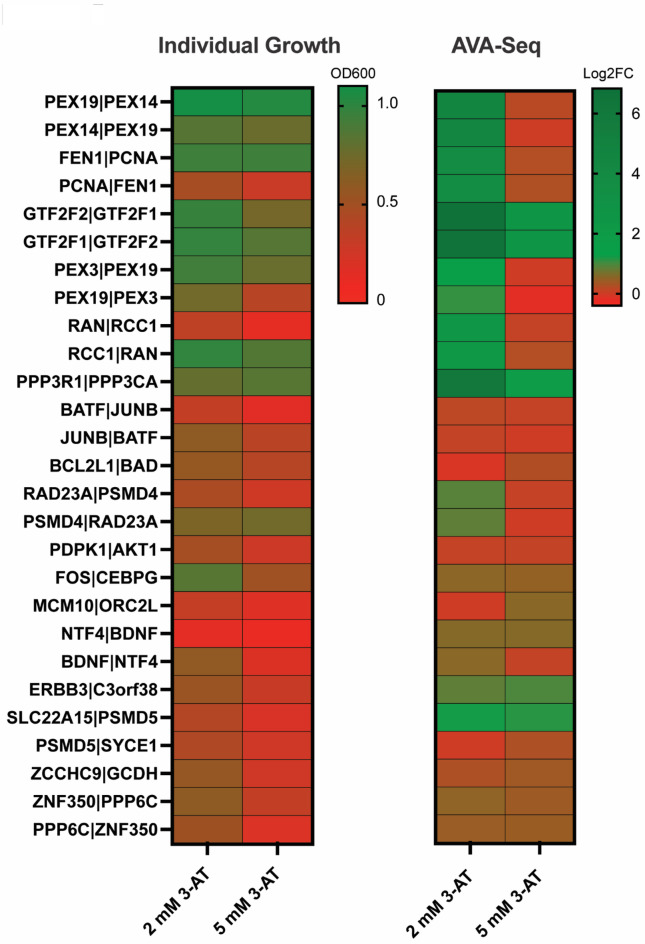
Given the increased predictions on interactome size and demand for protein function information, methods for detecting protein-protein interactions remain a significant development area. The all-vs.-all sequencing (AVA-Seq) method utilizes a convergent fusion plasmid design to make two-hybrid technology amenable to next-generation sequencing. Here, we further innovate to take advantage of synthetic DNA technologies and Oxford Nanopore Technologies long-read sequencing improvements to allow us to determine full-length protein-protein interactions. We tested 3,115 human protein-protein pairs using this approach and recovered 159 protein-protein interactions from a set of 57 full-length human proteins. Fifteen of the 159 full-length protein-protein interactions matched known human interactions. When referencing a human gold standard set of interactions, eight full-length protein-protein interactions were recovered from an expected 28 interaction pairs (28.6%), a typical recovery rate for two-hybrid technologies. The AVA-Seq method, in combination with the ease of synthetic DNA production and the MinION platform, offers a low-cost, high-throughput alternative for determining protein-protein interactions, which can be utilized in research labs at all stages.
Keywords: Long-read sequencing; Oxford nanopore technologies; Protein–protein interaction; Single-molecule sequencing; Two-hybrid.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures




References
-
- Weimann, M. et al. A Y2H-seq approach defines the human protein methyltransferase interactome. Nat. Methods10, 339–342 (2013). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
