

AChR Autoantibody Pathogenic Properties Are Heterogeneously Distributed and Undergo Temporal Changes Among Patients With Myasthenia Gravis
- PMID: 40680247
- PMCID: PMC12275905
- DOI: 10.1212/NXI.0000000000200436
AChR Autoantibody Pathogenic Properties Are Heterogeneously Distributed and Undergo Temporal Changes Among Patients With Myasthenia Gravis
Abstract
Background and objectives: Acetylcholine receptor (AChR) autoantibodies contribute to myasthenia gravis (MG) pathogenesis through 3 mechanisms: complement activation, receptor internalization, and acetylcholine (ACh) binding site blocking. Recently approved therapies target these autoantibodies by inhibiting the complement pathway or blocking the neonatal Fc receptor, reducing IgG autoantibody levels. However, these approaches have limitations: complement inhibitors do not address complement-independent mechanisms, and FcRn blockers only target IgG. Understanding how different pathogenic mechanisms, isotypes, and IgG subclasses are represented in the AChR autoantibody repertoire could lead to more precise application of therapeutics. To address this, we used advanced live cell-based assays to study autoantibody heterogeneity in longitudinally collected patient samples.
Methods: Serum samples (N = 210) from 50 AChR IgG+ generalized MG patients collected longitudinally over 2 years were evaluated using a set of cell-based assays to measure complement activation, receptor internalization, ACh binding site blocking, and the frequency of the IgM and IgA isotypes and IgG subclasses.
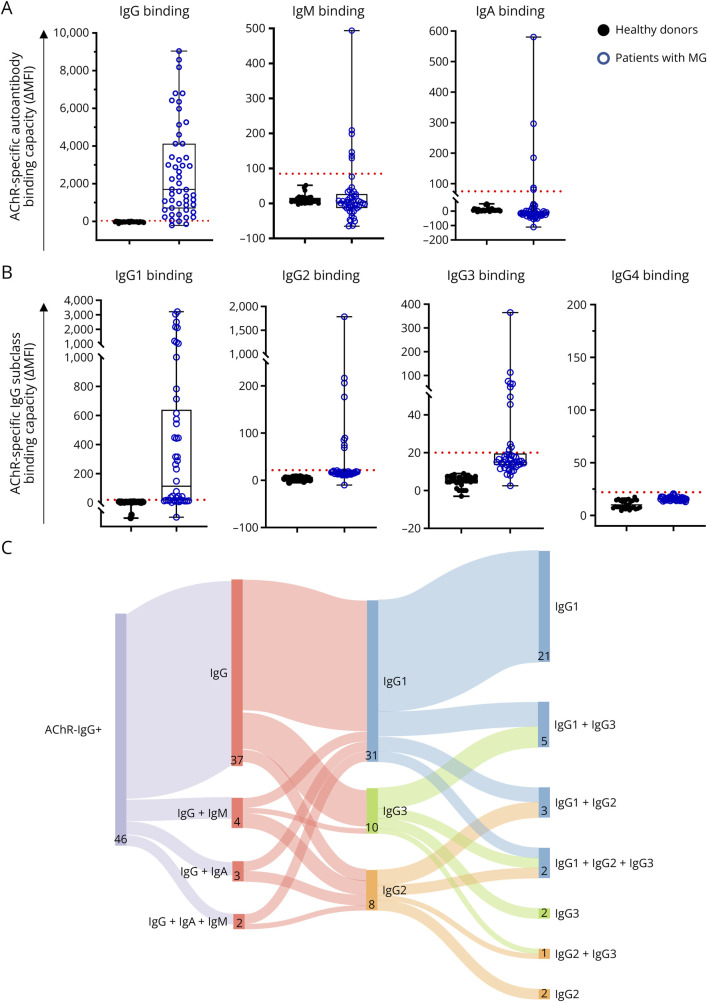
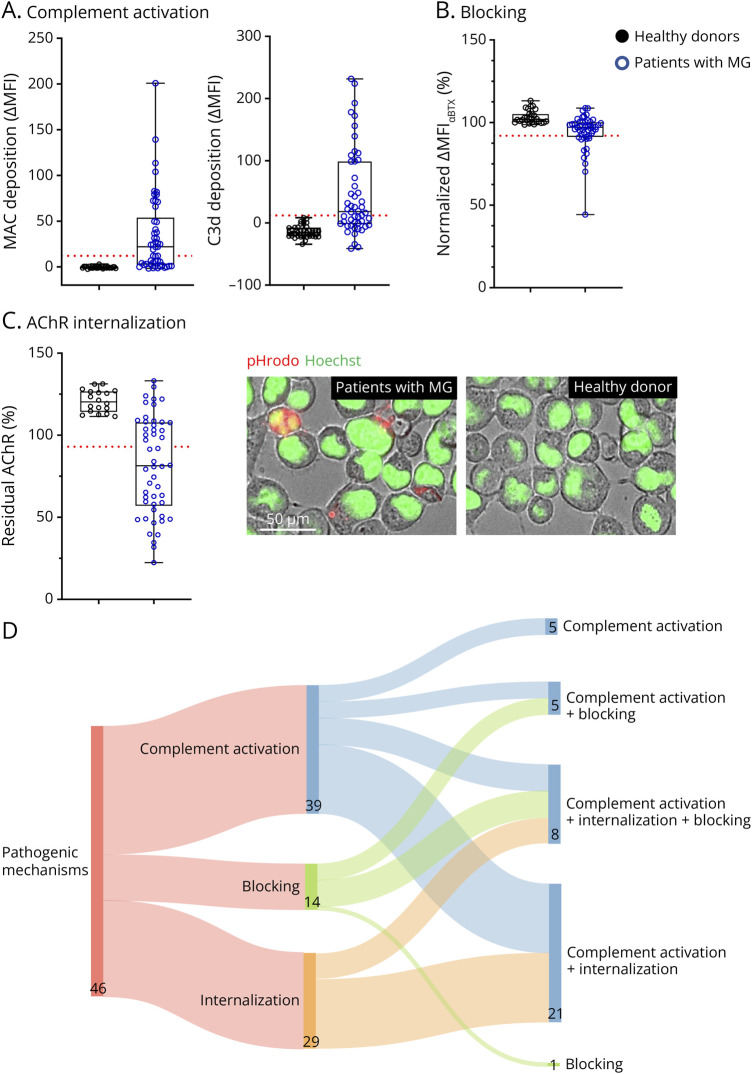
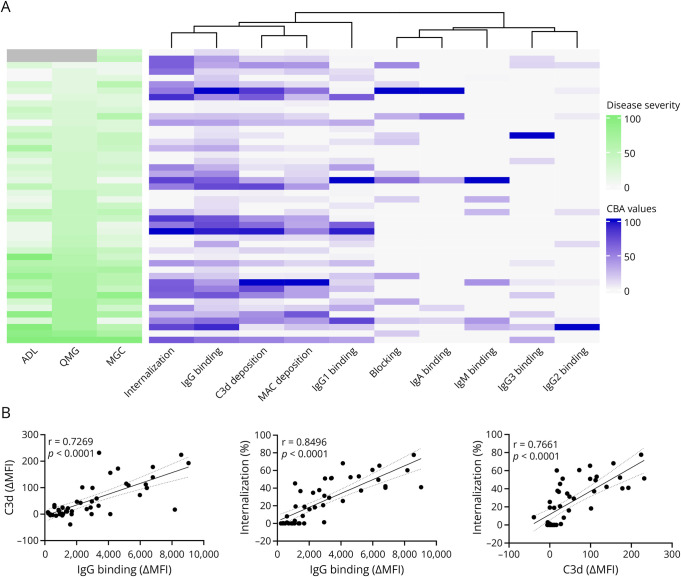
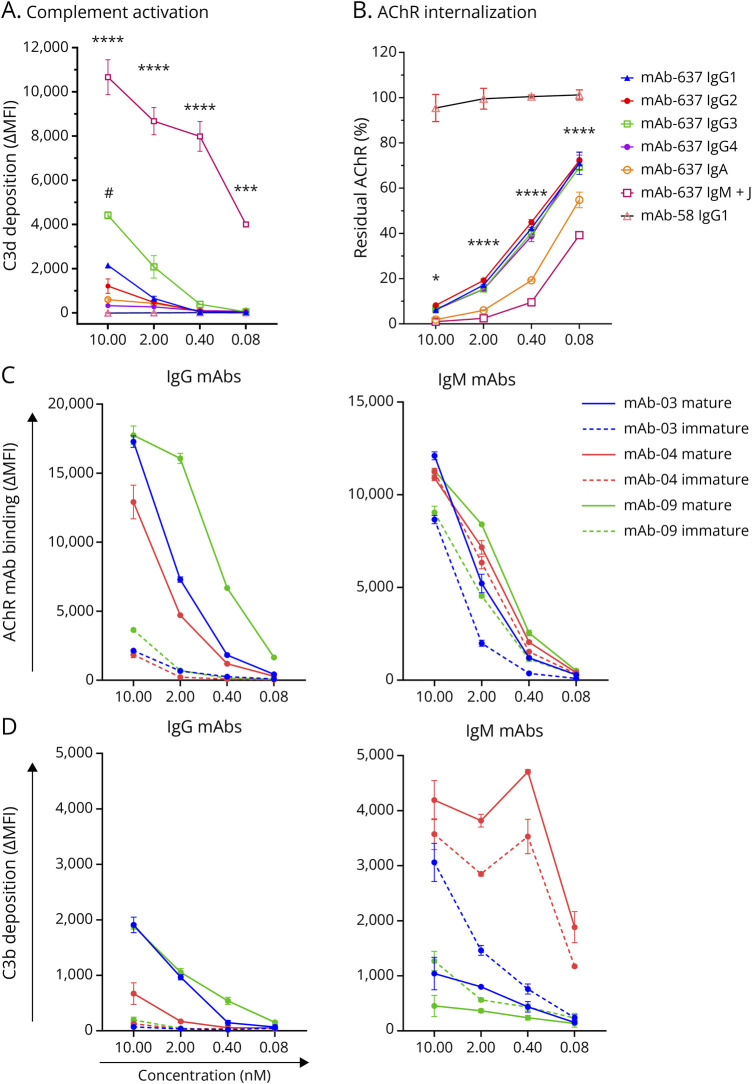
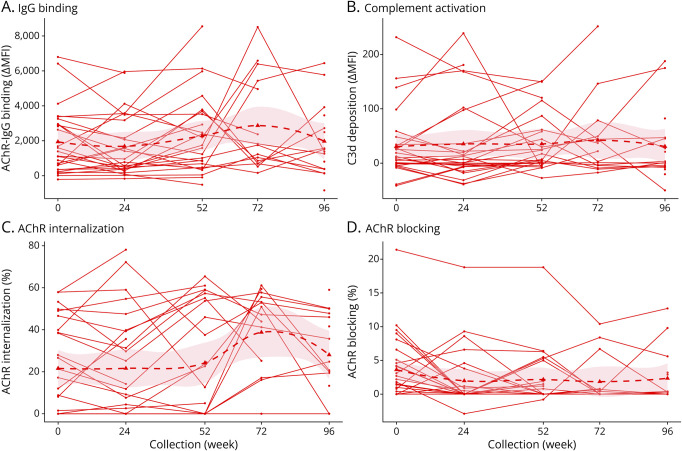
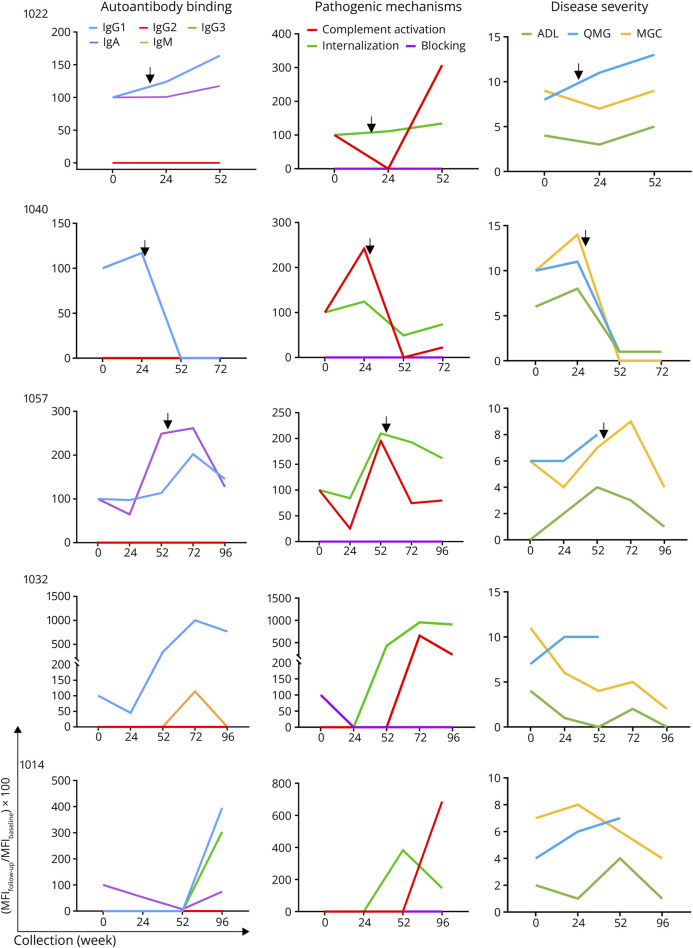
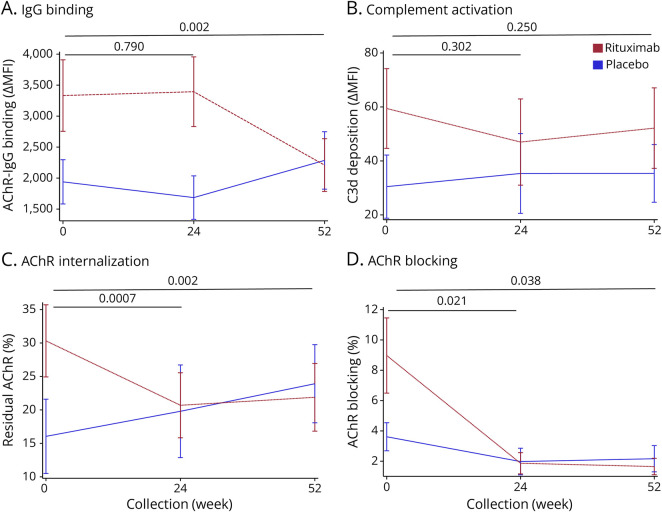
Results: In cross-sectional samples, IgA and IgM autoantibodies co-occurred with IgG in 10% and 12% of patients, respectively. In addition, 4% of patients had all 3 isotypes (IgA, IgM, and IgG) present simultaneously. AChR-IgG1 was found in 67.4%, followed by IgG3 (21.7%) and IgG2 (17.4%). Complement was active in 84.8%, followed by AChR internalization (63%) and blocking (30.4%). Complement and AChR internalization were simultaneously active in 45.6%, complement and blocking were active in 10.8%, and all 3 pathomechanisms were active in 17.4%. Blocking alone was active in only 2.1%; AChR internalization alone was not found. Autoantibody binding capacity was associated with the magnitude of complement activation and AChR internalization. Temporal fluctuations of autoantibody binding capacity and the associated pathogenic mechanisms were observed. Pathogenic mechanisms were not associated with disease severity in cross-sectional analyses. However, longitudinally, disease severity measures followed a similar trend to the AChR autoantibody repertoire and mediated pathogenic mechanisms in some individuals, but not others.
Discussion: These findings highlight subsets of patients with MG with autoantibodies that can mediate pathogenic mechanisms or include isotypes that some therapeutics may not effectively target. Consequently, we suggest incorporating comprehensive autoantibody profiling into future MG clinical trials to further investigate potential associations with treatment outcomes.
Conflict of interest statement
K.C.O. has received research support from Ra Pharma, now (UCB Pharma), Alexion Rare Disease (Astra Zeneca), Viela Bio (Horizon Therapeutics/Amgen), argenx, and Seismic Therapeutic. K.C.O. is an equity shareholder of Cabaletta Bio. K.C.O. has served on advisory boards for Roche, Merck (EMD Serono), and IgM Biosciences, and received speaking fees from Amgen and argenx. B.R. has been a consultant/advisor for Alexion (now part of AstraZeneca), Takeda, and argenx. In addition, B.R. has received research support from the Martin Shubik Fund for IBM at Yale University, NIH, Abcuro Pharmaceuticals, Immunovant, Takeda. B.R. is a shareholder of Cabaletta Bio. R.J.N. has received research support from the NIH, Genentech, Alexion (Astra Zeneca), argenx, Annexon Biosciences, Ra Pharmaceuticals (now UCB), MGFA , Momenta (now Janssen), Immunovant, Grifols, and Viela Bio (Horizon Therapeutics, now Amgen). R.J.N. has also served as a consultant/advisor for Alexion (Astra Zeneca), argenx, Cabaletta Bio, CSL Behring, Grifols, Ra Pharmaceuticals (now UCB Pharma), Immunovant, Momenta (now Janssen), Viela Bio (Horizon Therapeutics, now Amgen). The authors have no additional financial interests. All other authors declare no competing financial interests. Go to
Figures
Similar articles
-
Heterogeneity of Acetylcholine Receptor Autoantibody-Mediated Complement Activity in Patients With Myasthenia Gravis.Neurol Neuroimmunol Neuroinflamm. 2022 Apr 26;9(4):e1169. doi: 10.1212/NXI.0000000000001169. Print 2022 Jul. Neurol Neuroimmunol Neuroinflamm. 2022. PMID: 35473886 Free PMC article.
-
Autoantibodies in myasthenia gravis.Int Rev Neurobiol. 2025;182:89-119. doi: 10.1016/bs.irn.2025.04.024. Epub 2025 Jul 4. Int Rev Neurobiol. 2025. PMID: 40675742 Review.
-
Unknown Immunoregulatory Effects of FcRn Inhibition by Efgartigimod in Myasthenia Gravis: A New Mechanism of Action Beyond IgG Reduction.Neurol Neuroimmunol Neuroinflamm. 2025 Sep;12(5):e200455. doi: 10.1212/NXI.0000000000200455. Epub 2025 Jul 31. Neurol Neuroimmunol Neuroinflamm. 2025. PMID: 40743488 Free PMC article.
-
Association of blocking acetylcholine receptor antibodies with clinical subtypes and disease severity in myasthenia gravis.Clin Chim Acta. 2025 Sep 1;577:120449. doi: 10.1016/j.cca.2025.120449. Epub 2025 Jun 25. Clin Chim Acta. 2025. PMID: 40578418
-
The Black Book of Psychotropic Dosing and Monitoring.Psychopharmacol Bull. 2024 Jul 8;54(3):8-59. Psychopharmacol Bull. 2024. PMID: 38993656 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous