

Integrative single-cell and spatial transcriptomics uncover ELK4-mediated mechanisms in NDUFAB1+ tumor cells driving gastric cancer progression, metabolic reprogramming, and immune evasion
- PMID: 40688093
- PMCID: PMC12271198
- DOI: 10.3389/fimmu.2025.1591123
Integrative single-cell and spatial transcriptomics uncover ELK4-mediated mechanisms in NDUFAB1+ tumor cells driving gastric cancer progression, metabolic reprogramming, and immune evasion
Abstract
Background: Globally, gastric cancer (GC) stands as the fifth most prevalent form of malignant neoplasm and represents a significant contributor to mortality associated with oncological conditions. Despite advancements in therapeutic strategies for GC, the outcomes for patients with advanced stages of the disease continue to be unfavorable, largely due to tumor heterogeneity and the challenges posed by resistance to therapeutic agents. Metabolic reprogramming is pivotal in driving the advancement of GC, contributing to the development of resistance to pharmacological treatments and facilitating the cancer's ability to evade immune surveillance. Developing multi-target comprehensive treatment strategies by integrating tumor microenvironment (TME) modulation holds promise for significantly improving therapeutic efficacy.
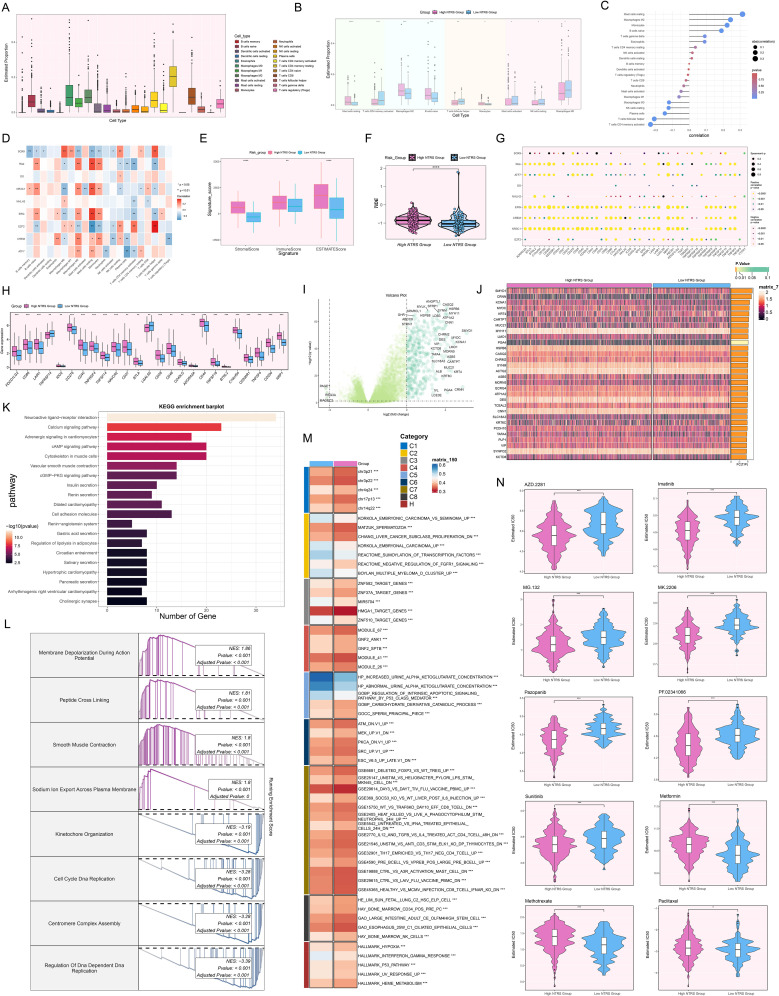
Methods: The study analyzed GC and identified key cell subtypes by integrating data derived from single-cell RNA-sequencing (scRNA-seq) alongside spatial transcriptomics information. Cell type identification was accomplished using the tool of Seurat, and the spatial distribution of cell types was revealed through the Robust Cell Type Decomposition technique. CellChat was used to analyze the interactions between key cell subtypes and other cells, and the "StLearn" package was employed to investigate spatial cell communication in depth. Additionally, the functional role of the key molecule ELK4 was validated through in vitro experiments.
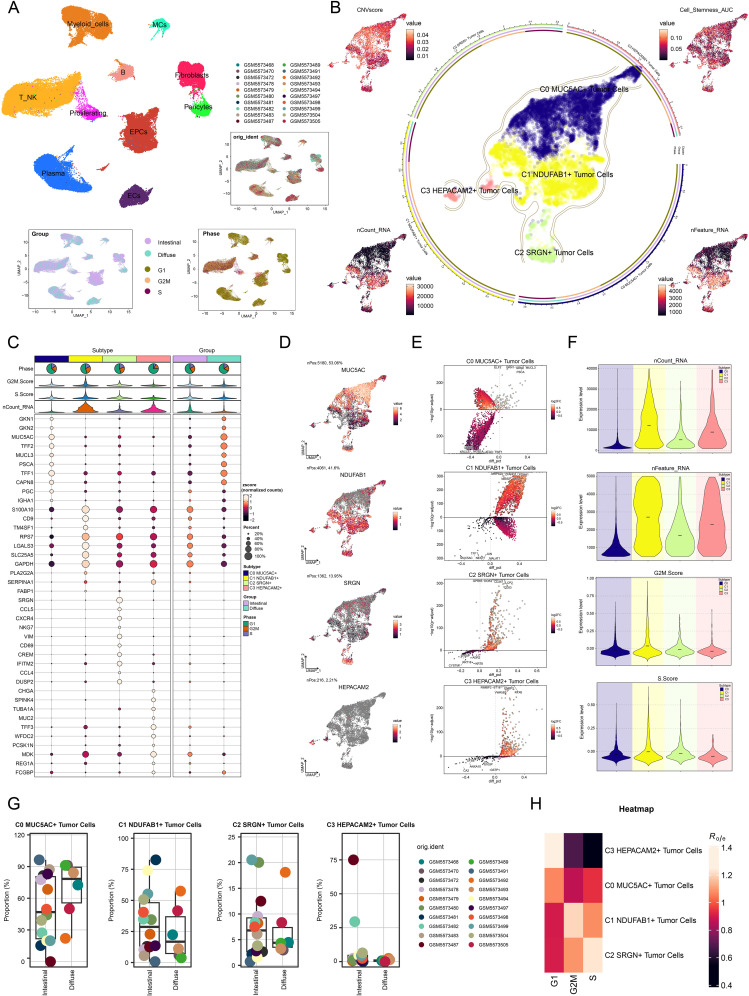
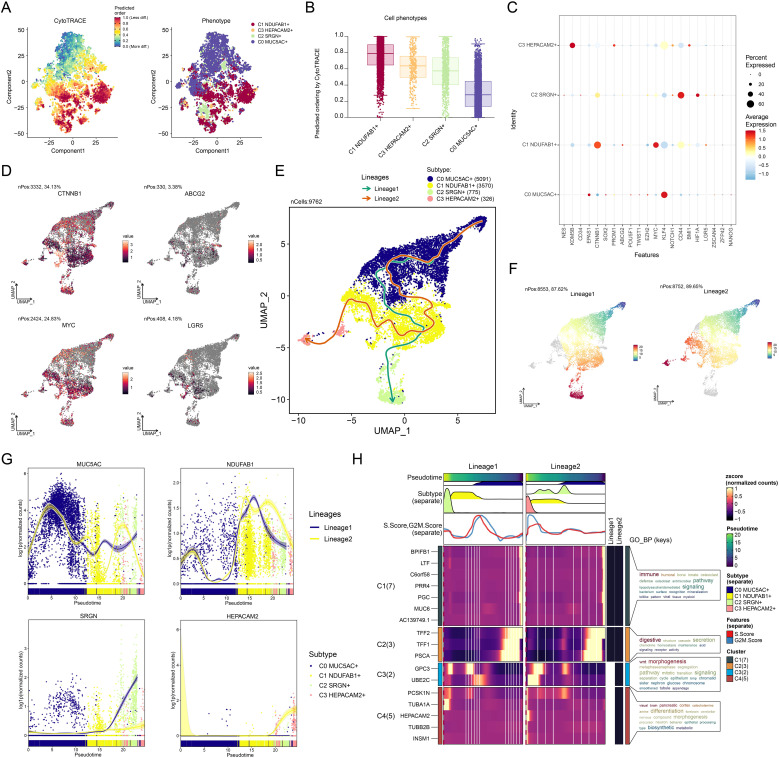
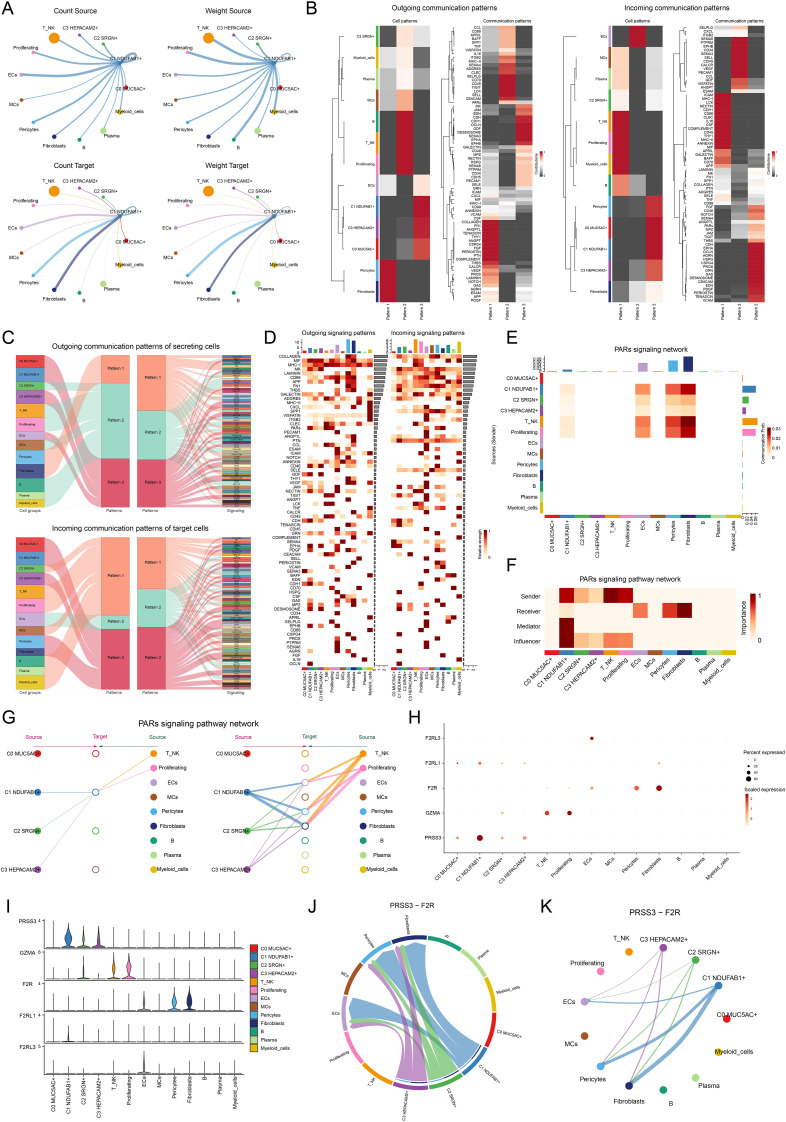
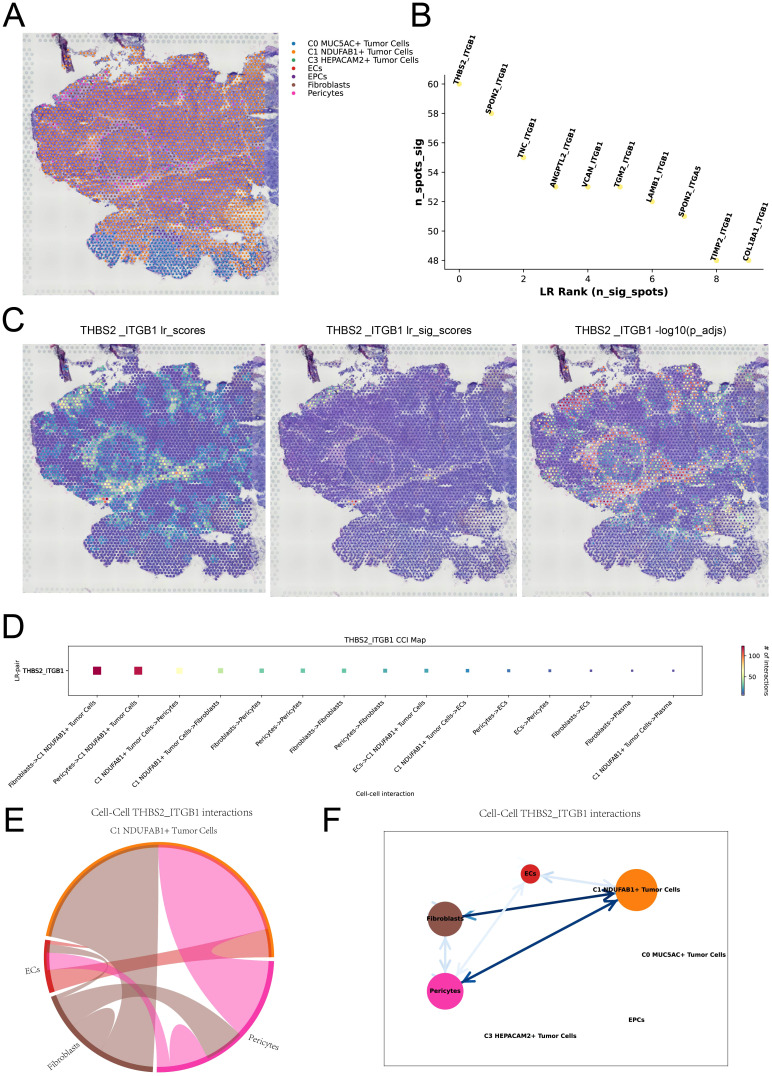
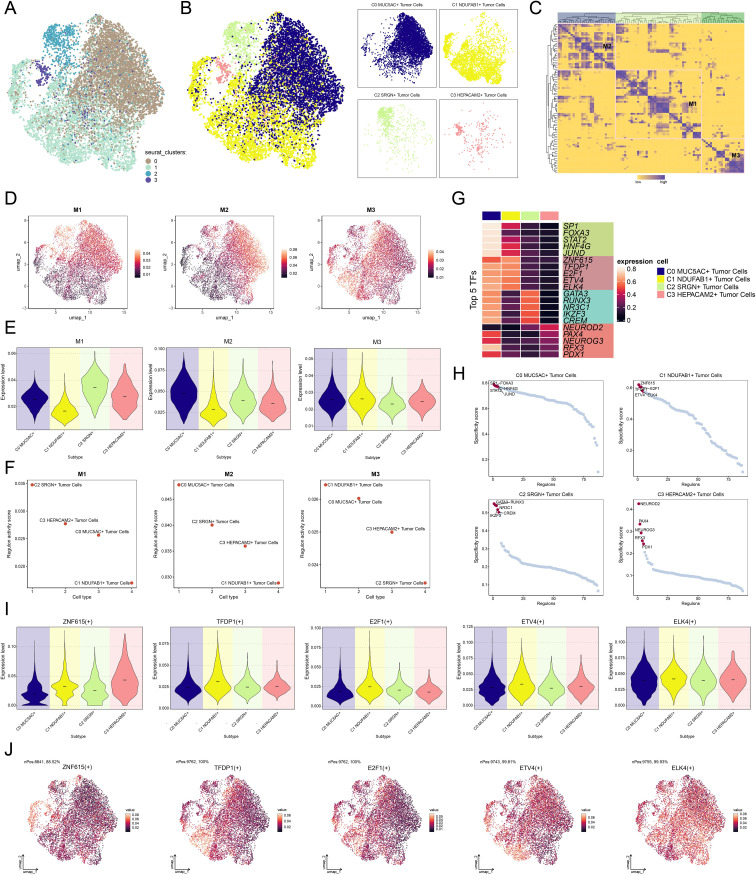
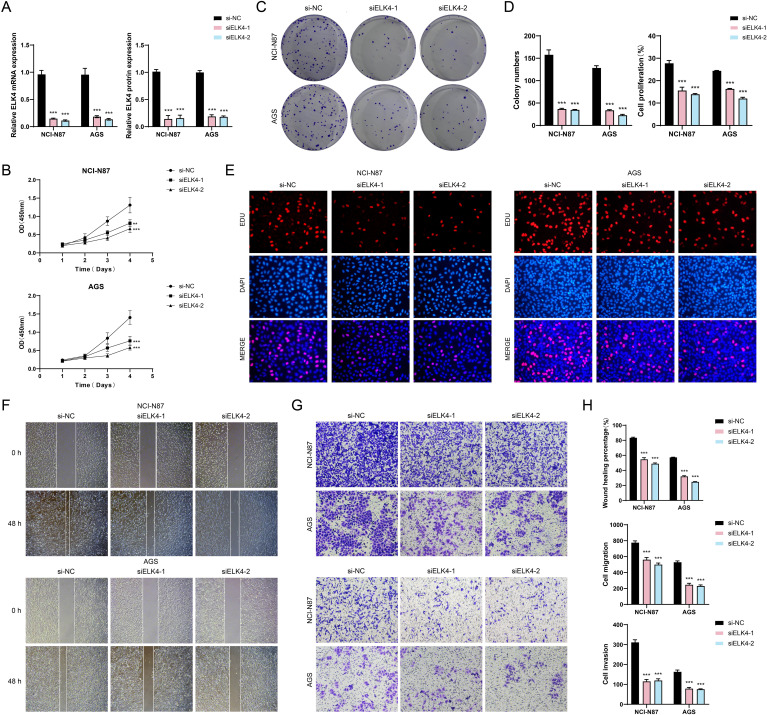
Results: This research utilized scRNA-seq combined with spatial transcriptomics to comprehensively analyze GC, identifying the C1 NDUFAB1+ subtype, which exhibited high proliferative activity, metabolic reprogramming capabilities, and immune evasion properties. It was found that the C1 NDUFAB1+ subtype closely interacted with fibroblasts and pericytes via the PARs signaling pathway. Additionally, in vitro experiments confirmed that knockdown of ELK4 substantially curbed tumor cell proliferation, migration, and invasion.
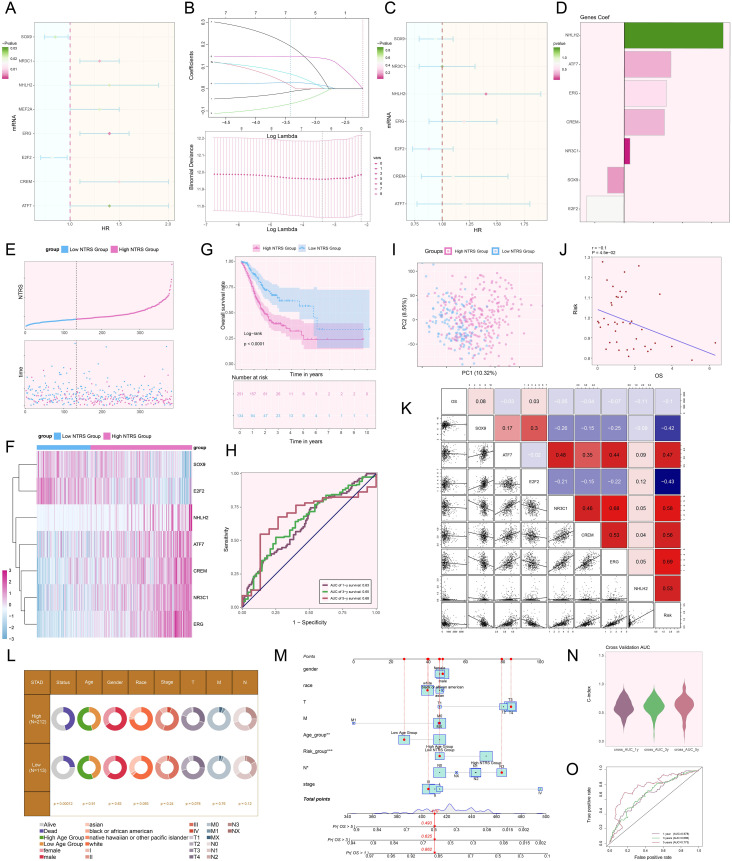
Conclusion: This study revealed the main significance of the C1 NDUFAB1+ subtype in GC, elucidating its core mechanisms in tumor progression, metabolic reprogramming, and immune evasion. ELK4 was identified as a key regulatory factor that markedly enhanced the proliferation, migratory capacity, and invasive potential of tumor cells, while changes in the TME were a driving force behind immune suppression and drug resistance. The findings underscored the importance of developing specific therapeutic targets, targeting metabolic reprogramming, and overcoming immune evasion, providing new theoretical foundations.
Keywords: gastric cancer; immune evasion; metabolic reprogramming; scRNA-seq; spatial transcriptomics; tumor microenvironment.
Copyright © 2025 Sun, Nie, Xiahou, Wang, Liu, Liu, Lin and Liu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Integrated single-cell and transcriptomic analysis of bone marrow-derived metastatic neuroblastoma reveals molecular mechanisms of metabolic reprogramming.Sci Rep. 2025 Aug 5;15(1):28519. doi: 10.1038/s41598-025-13626-8. Sci Rep. 2025. PMID: 40764361 Free PMC article.
-
Single-cell and spatial atlas of glioblastoma heterogeneity: characterizing the PCLAF+ subtype and YEATS4's oncogenic role.Front Immunol. 2025 Jul 25;16:1614549. doi: 10.3389/fimmu.2025.1614549. eCollection 2025. Front Immunol. 2025. PMID: 40787449 Free PMC article.
-
Integrated transcriptomics and machine learning reveal REN as a dual regulator of tumor stemness and NK cell evasion in Wilms tumor progression.Front Immunol. 2025 Jun 4;16:1612987. doi: 10.3389/fimmu.2025.1612987. eCollection 2025. Front Immunol. 2025. PMID: 40534868 Free PMC article.
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
-
Diagnostic accuracy of endoscopic ultrasonography (EUS) for the preoperative locoregional staging of primary gastric cancer.Cochrane Database Syst Rev. 2015 Feb 6;2015(2):CD009944. doi: 10.1002/14651858.CD009944.pub2. Cochrane Database Syst Rev. 2015. PMID: 25914908 Free PMC article.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous