

Association between fumarate hydratase variant subtypes and the risk of HLRCC-associated renal cell carcinoma: systematic review and meta-analysis
- PMID: 40691606
- PMCID: PMC12282009
- DOI: 10.1186/s40246-025-00797-8
Association between fumarate hydratase variant subtypes and the risk of HLRCC-associated renal cell carcinoma: systematic review and meta-analysis
Abstract
Background: Fumarate hydratase (FH) is a key mitochondrial enzyme in the tricarboxylic acid (TCA) cycle, catalyzing the reversible hydration of fumarate to malate, thereby facilitating aerobic ATP production and maintaining metabolic homeostasis. Germline pathogenic or likely pathogenic variants in the FH gene are strongly linked to hereditary leiomyomatosis and renal cell carcinoma (HLRCC), a rare hereditary cancer syndrome characterized by cutaneous and/or uterine leiomyomas and a markedly increased risk of renal cell carcinoma (RCC). These variants span a wide spectrum of genetic alterations, including missense, nonsense, frameshift, splice-site variants, as well as large genomic deletions. However, the relationship between specific pathogenic FH variant subtypes and the risk of developing HLRCC-associated RCC remains unclear. Therefore, this study systematically reviewed the existing literatures and conducted a meta-analysis to preliminarily explore the potential role of different functional subtypes of FH variants in the development of HLRCC-associated RCC, providing a basis for future clinical risk stratification and personalized surveillance strategies.
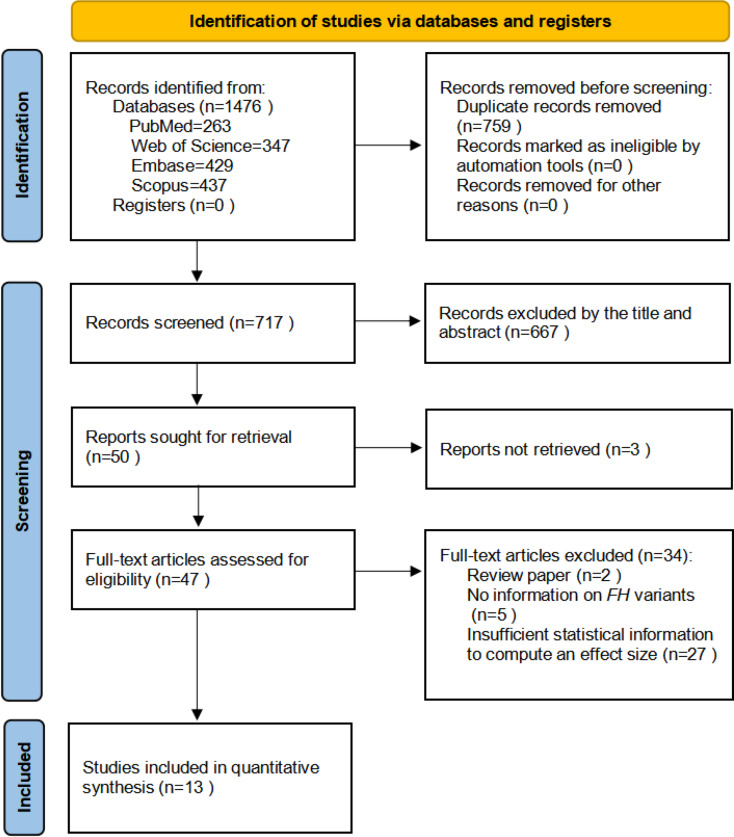
Methods: We systematically searched 4 major electronic databases—PubMed/MEDLINE, Embase, Scopus, and Web of Science—for relevant studies. To evaluate the association between pathogenic or likely pathogenic FH variant subtypes (Missense vs. Loss-of-Function (LOF)) and the risk of HLRCC-associated RCC, we performed a fixed-effects meta-analysis based on unadjusted odds ratios (ORs). In addition, exploratory subgroup analyses were performed by geographic region (North America and Europe), histological subtype (particularly type II papillary RCC (Type II PRCC)), tumor characteristics (such as distant metastasis and clinical stage), and study design (variant/gene-first vs. phenotype-first). These stratifications were intended to assess whether clinical or methodological features might modulate the observed associations and to provide context for future hypothesis-driven research. All statistical tests were two-sided, and heterogeneity was assessed using standard metrics. Effect estimates are reported as ORs with corresponding 95% confidence intervals (CIs).
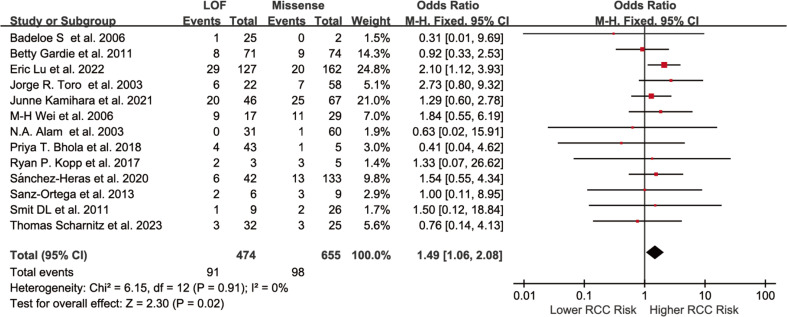
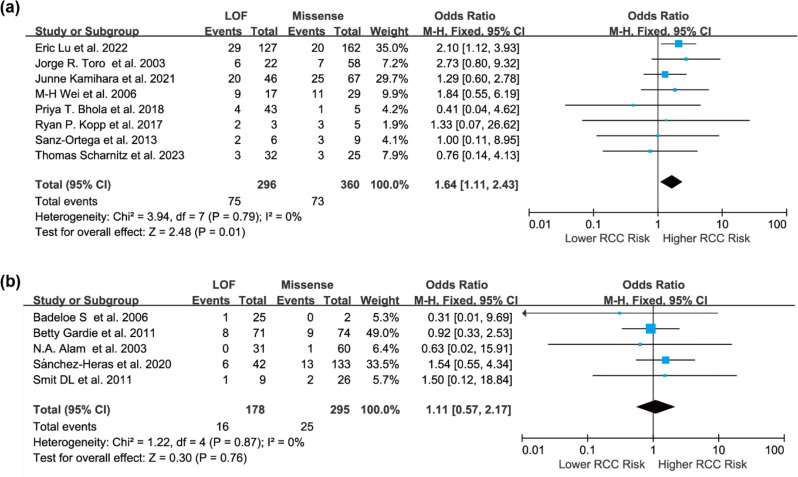
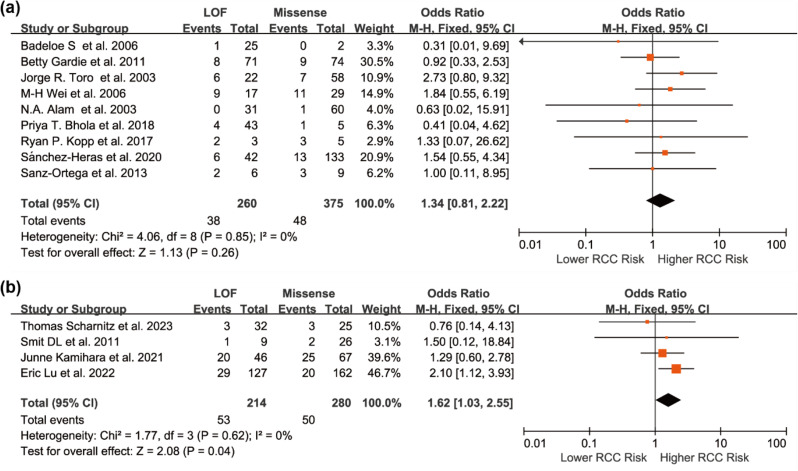
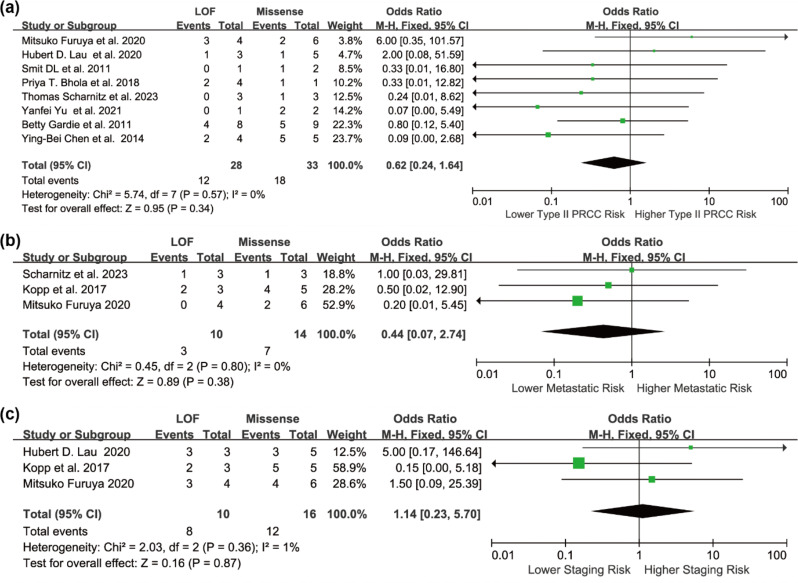
Results: Individuals harboring pathogenic or likely pathogenic FH LOF variants exhibited a significantly higher risk of developing HLRCC-associated RCC compared to those harboring missense variants (OR = 1.75, 95% CI: 1.28 to 2.38, p < 0.001). Subgroup analysis by geographic region showed a significant association in North American cohorts (OR = 1.64, 95% CI: 1.11 to 2.43, p < 0.05), while the association was not statistically significant in European cohorts (OR = 1.11, 95% CI: 0.57 to 2.17, p > 0.05). Stratification by study design further revealed a stronger association in variant-first or gene-first cohorts (OR = 1.62, 95% CI: 1.03 to 2.55, p < 0.05), while no significant association was observed in phenotype-first cohorts (OR = 1.34, 95% CI: 0.81 to 2.22, p > 0.05). Among patients diagnosed with HLRCC-associated RCC, those with LOF variants were more likely to present with advanced-stage disease at diagnosis. In contrast, patients with missense variants were more frequently associated with Type II PRCC and exhibited a higher propensity for distant metastasis.
Conclusion: This meta-analysis suggests that individuals harboring pathogenic or likely pathogenic FH LOF variants may have an approximately 1.75-fold higher risk of developing HLRCC-associated RCC compared to those with missense variants. While the pooled effect was statistically significant, subgroup analyses revealed regional and ascertainment-related differences, indicating potential underlying heterogeneity. These findings underscore the potential utility of FH variant subtypes as biomarkers for individualized risk assessment. Further prospective studies are warranted to validate these associations and guide surveillance strategies in hereditary renal cancer syndromes.
Supplementary Information: The online version contains supplementary material available at 10.1186/s40246-025-00797-8.
Conflict of interest statement
Declarations. Ethics, consent to participate, and consent to publish: Not applicable. Competing interests: The authors declare no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Miscellaneous
