

Increased ROS levels activate AMPK-ULK1-mediated mitophagy to promote pseudorabies virus replication
- PMID: 40691814
- PMCID: PMC12281776
- DOI: 10.1186/s13567-025-01595-9
Increased ROS levels activate AMPK-ULK1-mediated mitophagy to promote pseudorabies virus replication
Abstract
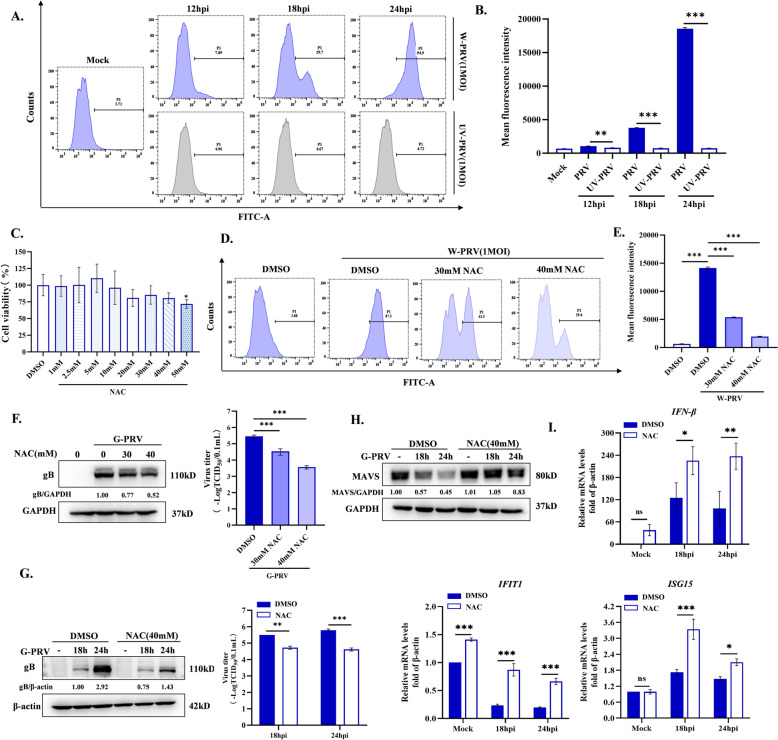
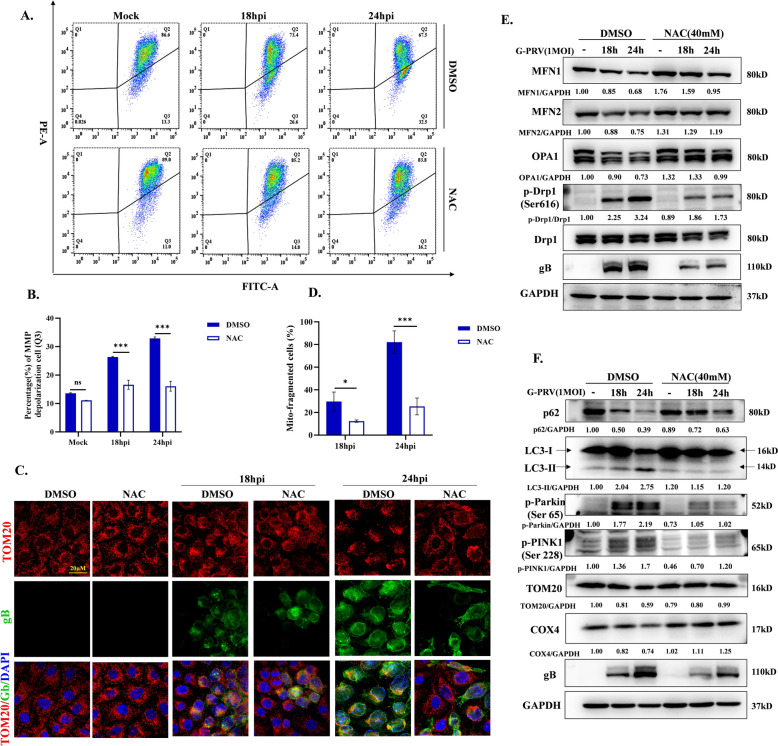
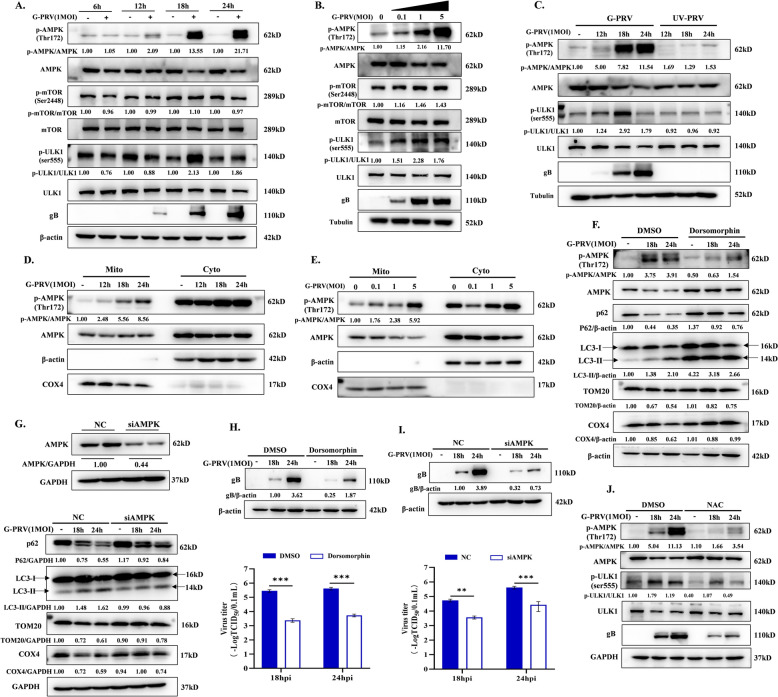
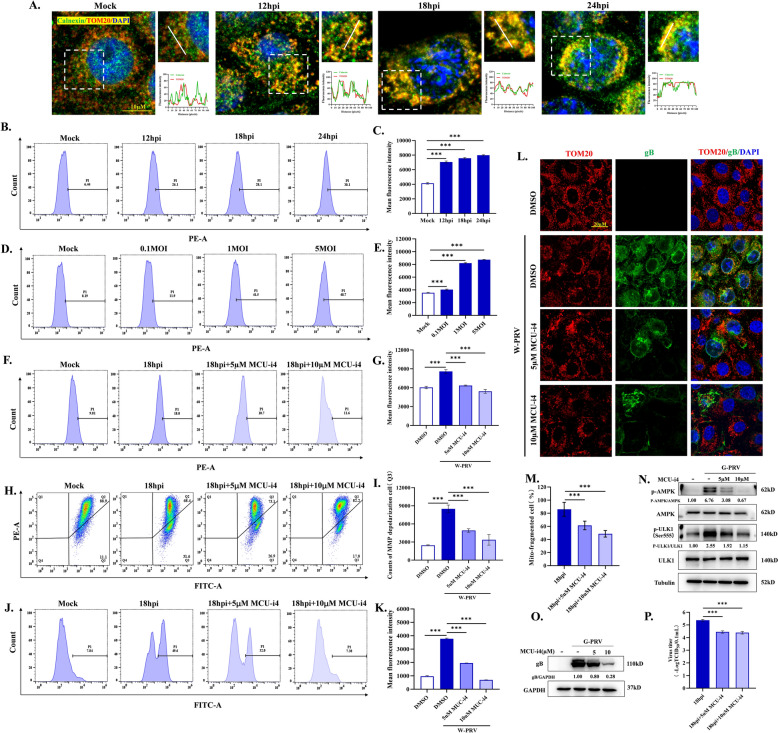
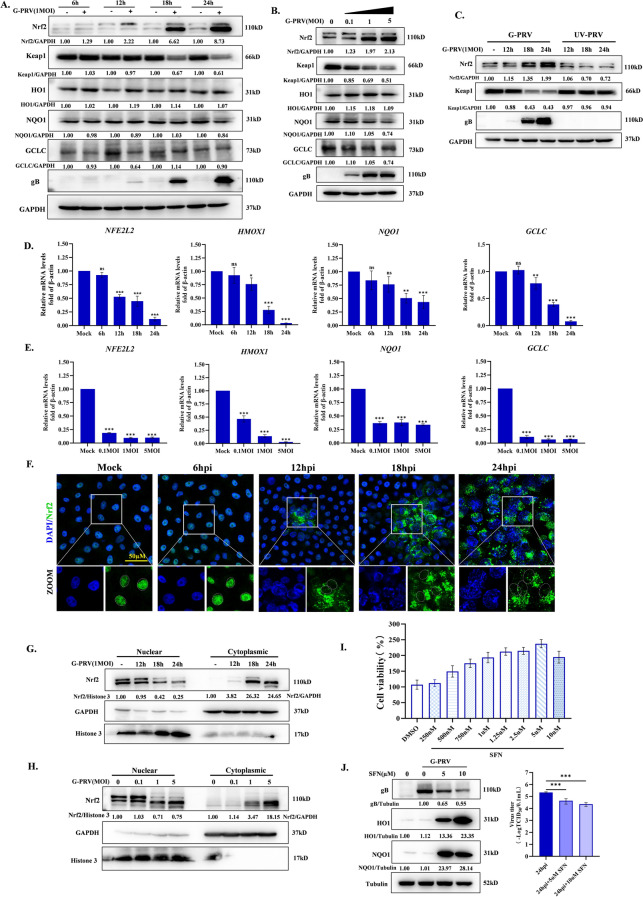
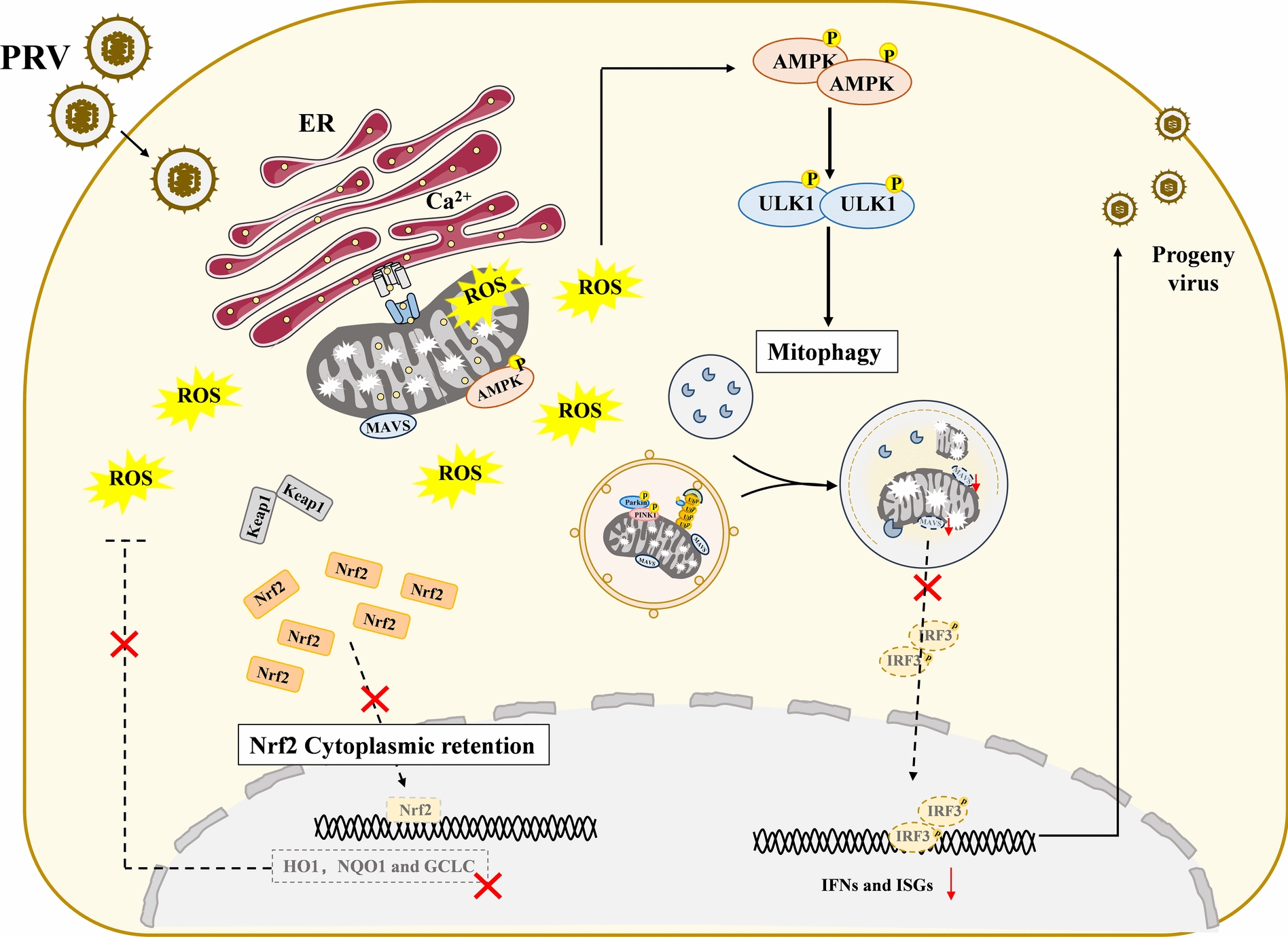
Increasing evidence has confirmed that oxidative stress plays a nonnegligible role in the viral pathogenic process. In this study, we investigated the role of reactive oxygen species (ROS) in the replication of pseudorabies virus (PRV). Our data showed that PRV infection initially enhanced the contact between the endoplasmic reticulum (ER) and mitochondria, leading to an upsurge of mitochondrial Ca2+ (mtCa2+) concentration, which resulted in the loss of mitochondrial membrane potential (MMP) and excessive ROS production. Instead of translocating it to the nucleus, PRV infection concurrently sequestered Nrf2 in cytoplasm impeding the efficient scavenging of intracellular ROS. The excessive ROS production and failure in ROS clearance contributed to the persistently high ROS levels during PRV infection. Furthermore, elevated ROS levels elicited activation of the AMPK-ULK1 axis, initiating PINK1-Parkin-dependent mitophagy that selectively degraded damaged mitochondria along with mitochondrial-localized mitochondrial antiviral signaling protein (MAVS). This process suppressed MAVS-mediated type I interferon responses by eliminating both dysfunctional mitochondria and their associated antiviral signaling platforms, thereby creating a cellular environment permissive to viral replication. Overall, our findings elucidated the mechanism by which ROS enables the virus to resist the host interferon immune response and provided a theoretical basis for ROS-based antiviral strategies.
Keywords: AMPK; Nrf2; Pseudorabies virus; mitochondria Ca2+; mitophagy; reactive oxygen species.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare that they have no competing interests.
Figures
Similar articles
-
Pseudorabies virus infection triggers mitophagy to dampen the interferon response and promote viral replication.J Virol. 2024 Oct 22;98(10):e0104824. doi: 10.1128/jvi.01048-24. Epub 2024 Aug 30. J Virol. 2024. PMID: 39212384 Free PMC article.
-
Role of AMBRA1 in mitophagy regulation: emerging evidence in aging-related diseases.Autophagy. 2024 Dec;20(12):2602-2615. doi: 10.1080/15548627.2024.2389474. Epub 2024 Sep 2. Autophagy. 2024. PMID: 39113560 Free PMC article. Review.
-
Sirt3 Protects Retinal Pigment Epithelial Cells From High Glucose-Induced Injury by Promoting Mitophagy Through the AMPK/mTOR/ULK1 Pathway.Transl Vis Sci Technol. 2024 Mar 1;13(3):19. doi: 10.1167/tvst.13.3.19. Transl Vis Sci Technol. 2024. PMID: 38517447 Free PMC article.
-
Antiviral activity and antioxidant activity of Cistanche deserticola polysaccharides against pseudorabies viruses.Int J Biol Macromol. 2025 Sep;321(Pt 2):146326. doi: 10.1016/j.ijbiomac.2025.146326. Epub 2025 Jul 25. Int J Biol Macromol. 2025. PMID: 40716544
-
Copper dyshomeostasis and its relationship to AMPK activation, mitochondrial dynamics, and biogenesis of mitochondria: A systematic review of in vivo studies.J Trace Elem Med Biol. 2024 Dec;86:127549. doi: 10.1016/j.jtemb.2024.127549. Epub 2024 Oct 11. J Trace Elem Med Biol. 2024. PMID: 39427561
References
-
- Nencioni L, Sgarbanti R, De Chiara G, Garaci E, Palamara AT (2007) Influenza virus and redox mediated cell signaling: a complex network of virus/host interaction. New Microbiol 30:367–375 - PubMed
-
- Protto V, Tramutola A, Fabiani M, Marcocci ME, Napoletani G, Iavarone F, Vincenzoni F, Castagnola M, Perluigi M, Di Domenico F, De Chiara G, Palamara AT (2020) Multiple herpes simplex virus-1 (HSV-1) reactivations induce protein oxidative damage in mouse brain: novel mechanisms for Alzheimer’s disease progression. Microorganisms 8:972 - PMC - PubMed
-
- Hu J, Li H, Luo X, Li Y, Bode A, Cao Y (2017) The role of oxidative stress in EBV lytic reactivation, radioresistance and the potential preventive and therapeutic implications. Int J Cancer 141:1722–1729 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
