

Description and phenotype of a novel C5 gene mutation and a novel combination: family report and literature review
- PMID: 40692790
- PMCID: PMC12277378
- DOI: 10.3389/fimmu.2025.1605903
Description and phenotype of a novel C5 gene mutation and a novel combination: family report and literature review
Abstract
Background: Patients with C5 mutations are more susceptibility to Gram-negative bacterial infections, particularly Neisseria species.
Objective: To describe the phenotype and clinical features of a family carrying two C5 gene variants, including one novel mutation, and to assess their functional and genetic significance.
Methods: We analyzed the clinical and genetic characteristics of a family with two compounds heterozygous C5 variants. Clinical features were assessed across affected and unaffected family members, and results were correlated with genetic and functional assays.
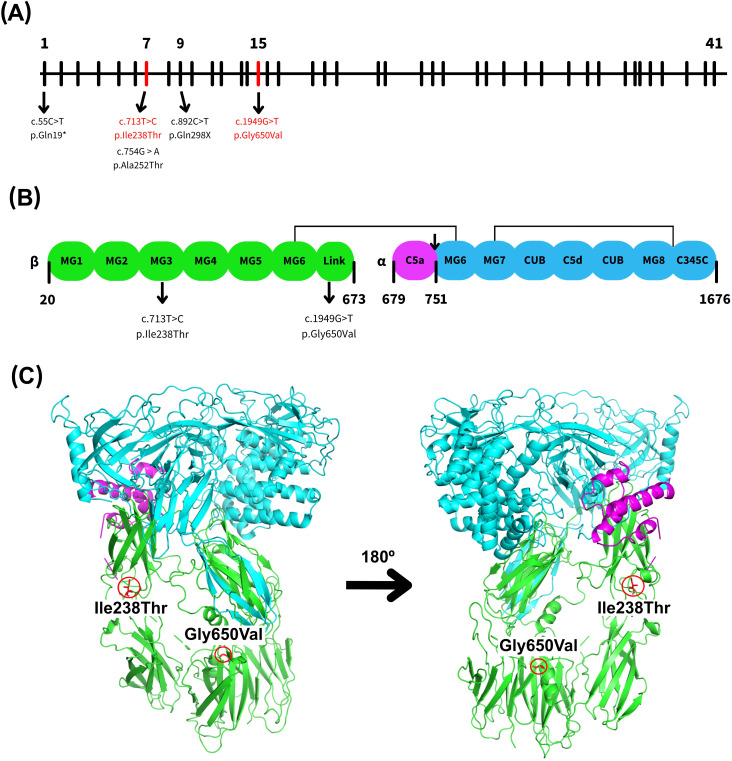
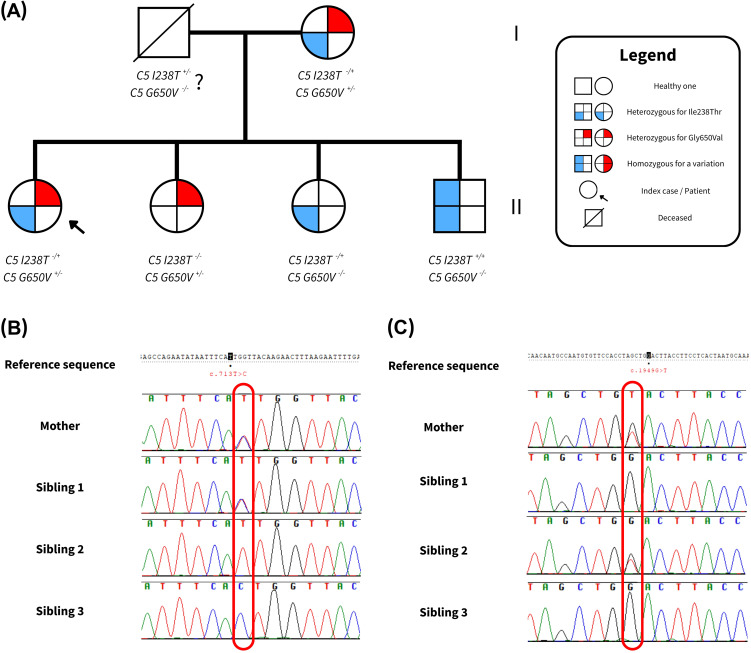
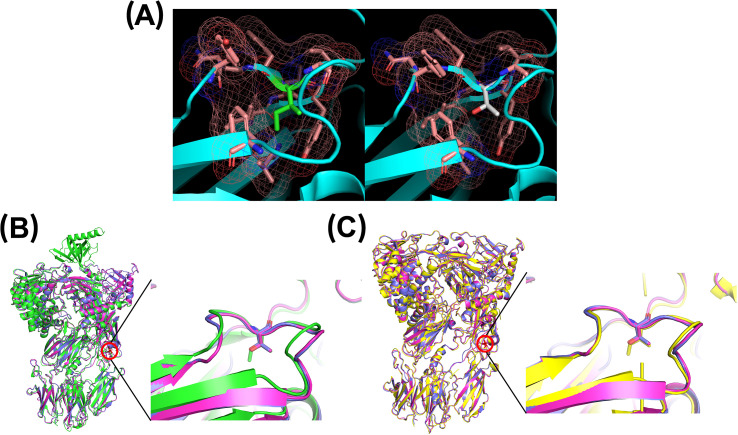
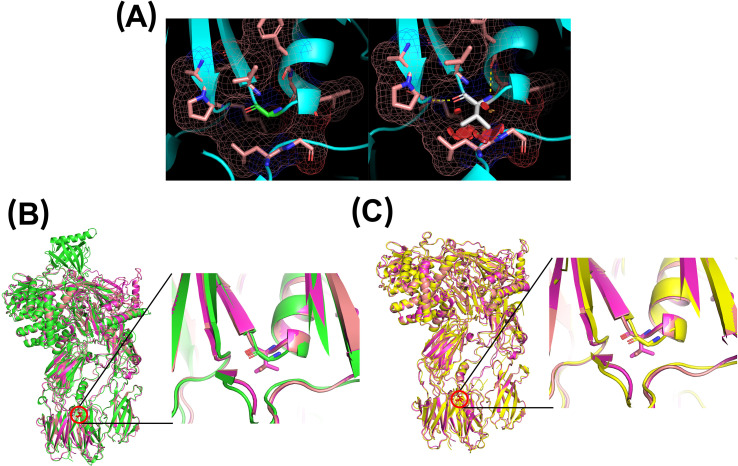
Results: Genetic testing revealed compound heterozygous variants in the C5 gene: c.713T>C (p.Ile238Thr) and c.1949G>T (p.Gly650Val). The p.Ile238Thr variant, located in exon 7, results in a substitution of isoleucine with threonine. The p.Gly650Val variant, located in exon 15, replaces glycine with valine. Sanger sequencing confirmed the variants were in trans (on separate alleles). The mother carried the same two variants as the patient. Two siblings carried one variant each (Gly650Val and Ile238Thr, respectively), and one sibling was homozygous for the Ile238Thr variant.Clinically, the patient, the mother, and the homozygous sibling had very low serum C5 protein and CH50 levels, correlating with increased susceptibility to Neisseria infections. Siblings carrying only one variant had normal complement function. In silico analysis and molecular modeling indicate that both amino acid substitutions (Ile238Thr and Gly650Val) may disrupt C5 protein structure. The Ile238Thr change introduces a polar residue in place of a hydrophobic one, disrupting the hydrophobic core and opening a loop between beta-sheets. The Gly650Val change substitutes a small residue with a larger one, causing steric hindrance that necessitates structural rearrangements, including shifts in a loop, alpha-helix, and beta-sheet.
Conclusion: We describe a novel C5 variant (Gly650Val) a previously reported variant (Ile238Thr) in unique genotypic combinations (compound heterozygous and homozygous) associated with marked C5 deficiency and increased susceptibility to invasive Neisseria infections. Our findings underscore the importance of combining genetic, functional, and structural data for variant interpretation in complement deficiencies.
Keywords: C5 deficiency; C5 mutation; complement deficiency; novel mutation; phenotype and genotype.
Copyright © 2025 Lizama-Muñoz, Gutiérrez-Bautista, Bernal and López-Nevot.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures
References
-
- Owen EP, Würzner R, Leisegang F, Rizkallah P, Whitelaw A, Simpson J, et al. A complement C5 gene mutation, c.754G>A:p.A252T, is common in the Western Cape, South Africa and found to be homozygous in seven percent of Black African meningococcal disease cases. Mol Immunol. (2015) 64:170–6. doi: 10.1016/J.MOLIMM.2014.11.010, PMID: - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous