

Integrative Single-Cell Analysis Reveals Iron Overload-Induced Senescence and Metabolic Reprogramming in Ovarian Endometriosis-Associated Infertility
- PMID: 40693455
- PMCID: PMC12362736
- DOI: 10.1002/advs.202417528
Integrative Single-Cell Analysis Reveals Iron Overload-Induced Senescence and Metabolic Reprogramming in Ovarian Endometriosis-Associated Infertility
Abstract
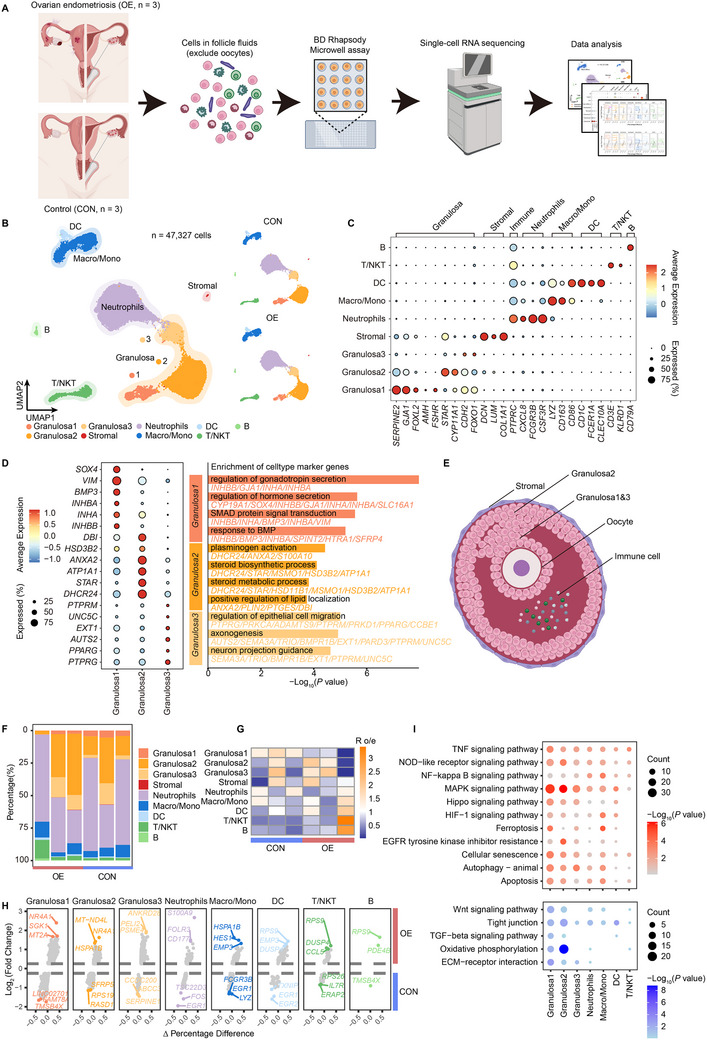
Endometriosis, particularly ovarian endometriosis (OE), is a major cause of infertility, often associated with reduced oocyte quality and impaired ovarian function. Iron overload plays a key role in OE progression. This study investigates the effects of iron overload on follicular function in OE-associated infertility (OEI). A single-cell atlas of pre-ovulatory follicular fluid from OEI patients reveals dynamic changes in iron metabolism and iron-induced senescence phenotypes. Spatial transcriptomics using Stereo-seq in iron-overloaded mouse ovaries further identifies localized senescence features. Additional analysis of aging human ovaries highlights conserved patterns of iron dysregulation. These findings provide mechanistic insight into iron overload-related ovarian pathology and suggest potential therapeutic targets for improving oocyte quality in OEI.
Keywords: endometriosis; infertility; iron; multi‐omics; senescence.
© 2025 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures







References
-
- Chapron C., Marcellin L., Borghese B., Santulli P., Nat. Rev. Endocrinol. 2019, 15, 666. - PubMed
-
- Taylor H. S., Kotlyar A. M., Flores V. A., Lancet 2021, 397, 839. - PubMed
-
- Giudice L. C., Kao L. C. E., Lancet 2004, 364, 1789. - PubMed
-
- Hamdan M., Dunselman G., Li T. C., Cheong Y., Hum. Reprod. Update 2015, 21, 809. - PubMed
MeSH terms
Substances
Grants and funding
- 82304962/National Natural Science Foundation of China
- 82074206/National Natural Science Foundation of China
- 82374503/National Natural Science Foundation of China
- 2024-2026QNRC001/the Young Elite Scientists Sponsorship Program by CAST
- ZCLQN25H2702/Zhejiang Provincial Natural Science Foundation of China
LinkOut - more resources
Full Text Sources
Medical