

Multilaboratory Untargeted Mass Spectrometry Metabolomics Collaboration to Identify Bottlenecks and Comprehensively Annotate A Single Dataset
- PMID: 40693864
- PMCID: PMC12332825
- DOI: 10.1021/acs.analchem.4c05577
Multilaboratory Untargeted Mass Spectrometry Metabolomics Collaboration to Identify Bottlenecks and Comprehensively Annotate A Single Dataset
Abstract
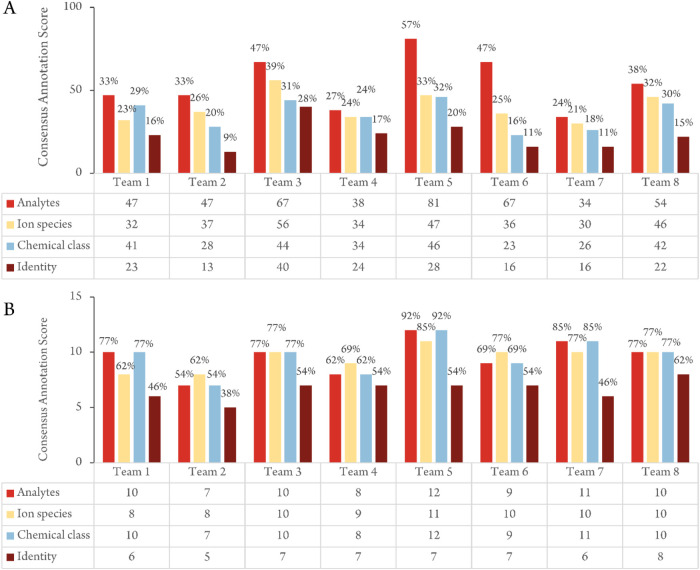
Annotation is the process of assigning features in mass spectrometry metabolomics data sets to putative chemical structures or "analytes." The purpose of this study was to identify challenges in the annotation of untargeted mass spectrometry metabolomics datasets and suggest strategies to overcome them. Toward this goal, we analyzed an extract of the plant ashwagandha (Withania somnifera) using liquid chromatography-mass spectrometry on two different platforms (an Orbitrap and Q-ToF) with various acquisition modes. The resulting 12 datasets were shared with ten teams that had established expertise in metabolomics data interpretation. Each team annotated at least one positive ion dataset using their own approaches. Eight teams selected the positive ion mode data-dependent acquisition (DDA) data collected on the Orbitrap platform, so the results reported for that dataset were chosen for an in-depth comparison. We compiled and cross-checked the annotations of this dataset from each laboratory to arrive at a "consensus annotation," which included 142 putative analytes, of which 13 were confirmed by comparison with standards. Each team only reported a subset (24 to 57%) of the analytes in the consensus list. Correct assignment of ion species (clusters and fragments) in MS spectra was a major bottleneck. In many cases, in-source redundant features were mistakenly considered to be independent analytes, causing annotation errors and resulting in overestimation of sample complexity. Our results suggest that better tools/approaches are needed to effectively assign feature identity, group related mass features, and query published spectral and taxonomic data when assigning putative analyte structures.
Figures



References
-
- Pezzatti J., Boccard J., Codesido S., Gagnebin Y., Joshi A., Picard D., Gonzalez-Ruiz V., Rudaz S.. Implementation of liquid chromatography-high resolution mass spectrometry methods for untargeted metabolomic analyses of biological samples: A tutorial. Anal. Chim. Acta. 2020;1105:28–44. doi: 10.1016/j.aca.2019.12.062. - DOI - PubMed
-
- Wolfender J.-L., Nuzillard J.-M., van der Hooft J. J. J., Renault J.-H., Bertrand S.. Accelerating Metabolite Identification in Natural Product Research: Toward an Ideal Combination of Liquid Chromatography–High-Resolution Tandem Mass Spectrometry and NMR Profiling, in Silico Databases, and Chemometrics. Anal. Chem. 2019;91(1):704–742. doi: 10.1021/acs.analchem.8b05112. - DOI - PubMed
-
- Sumner L. W., Amberg A., Barrett D., Beale M. H., Beger R., Daykin C. A., Fan T. W. M., Fiehn O., Goodacre R., Griffin J. L.. et al. Proposed minimum reporting standards for chemical analysis. Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI) Metabolomics. 2007;3(3):211–221. doi: 10.1007/s11306-007-0082-2. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
