

A novel expectation-maximization approach to infer general diploid selection from time-series genetic data
- PMID: 40694589
- PMCID: PMC12310050
- DOI: 10.1371/journal.pgen.1011769
A novel expectation-maximization approach to infer general diploid selection from time-series genetic data
Abstract
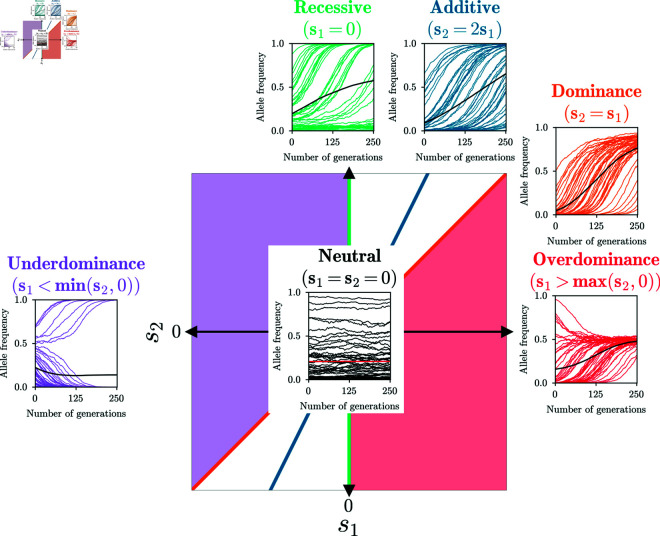
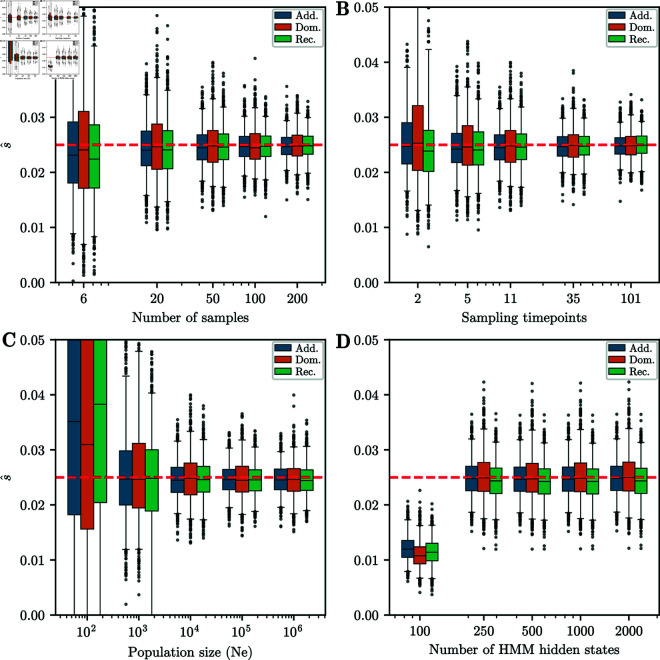
Detecting and quantifying the strength of selection is a major objective in population genetics. Since selection acts over multiple generations, many approaches have been developed to detect and quantify selection using genetic data sampled at multiple points in time. Such time-series genetic data is commonly analyzed using Hidden Markov Models, but in most cases, under the assumption of additive selection. However, many examples of genetic variation exhibiting non-additive mechanisms exist, making it critical to develop methods that can characterize selection in more general scenarios. Here, we extend a previously introduced expectation-maximization algorithm for the inference of additive selection coefficients to the case of general diploid selection, in which the heterozygote and homozygote fitness are parameterized independently. We furthermore introduce a framework to identify bespoke modes of diploid selection from given data, a heuristic to account for variable population size, and a procedure for aggregating data across linked loci to increase power and robustness. Using extensive simulation studies, we find that our method accurately and efficiently estimates selection coefficients for different modes of diploid selection across a wide range of scenarios; however, power to classify the mode of selection is low unless selection is very strong. We apply our method to ancient DNA samples from Great Britain in the last 4,450 years and detect evidence for selection in six genomic regions, including the well-characterized LCT locus. Our work is the first genome-wide scan characterizing signals of general diploid selection.
Copyright: © 2025 Fine, Steinrücken. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures













Update of
-
A novel expectation-maximization approach to infer general diploid selection from time-series genetic data.bioRxiv [Preprint]. 2025 May 26:2024.05.10.593575. doi: 10.1101/2024.05.10.593575. bioRxiv. 2025. Update in: PLoS Genet. 2025 Jul 22;21(7):e1011769. doi: 10.1371/journal.pgen.1011769. PMID: 38798346 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
