

Constructing a novel mitochondrial metabolism-related genes signature to evaluate tumor immune microenvironment and predict survival of colorectal cancer
- PMID: 40697926
- PMCID: PMC12279720
- DOI: 10.3389/fmed.2025.1618471
Constructing a novel mitochondrial metabolism-related genes signature to evaluate tumor immune microenvironment and predict survival of colorectal cancer
Abstract
Background: Colorectal cancer (CRC) is a highly lethal gastrointestinal malignancy with substantial global health implications. Although mitochondrial metabolism genes play a crucial role in CRC development, their prognostic significance remains unclear.
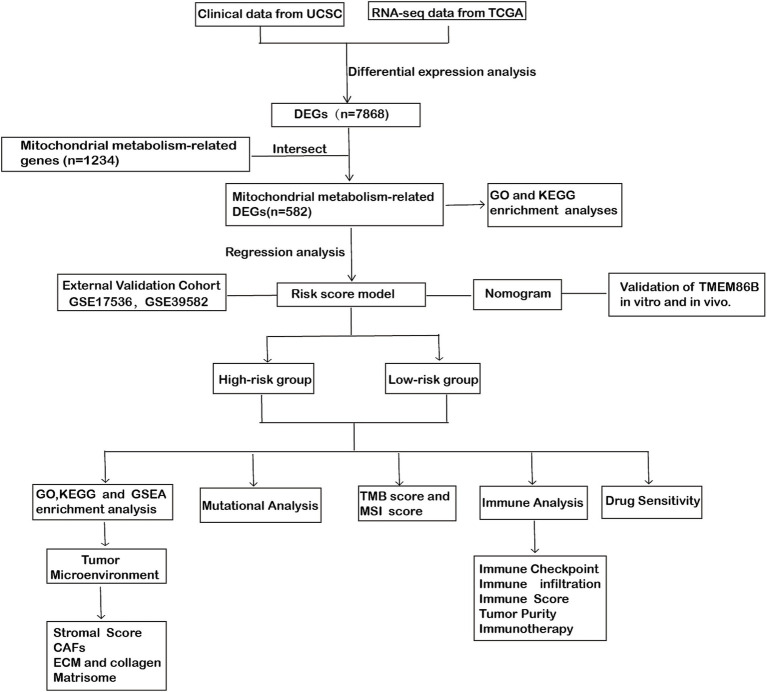
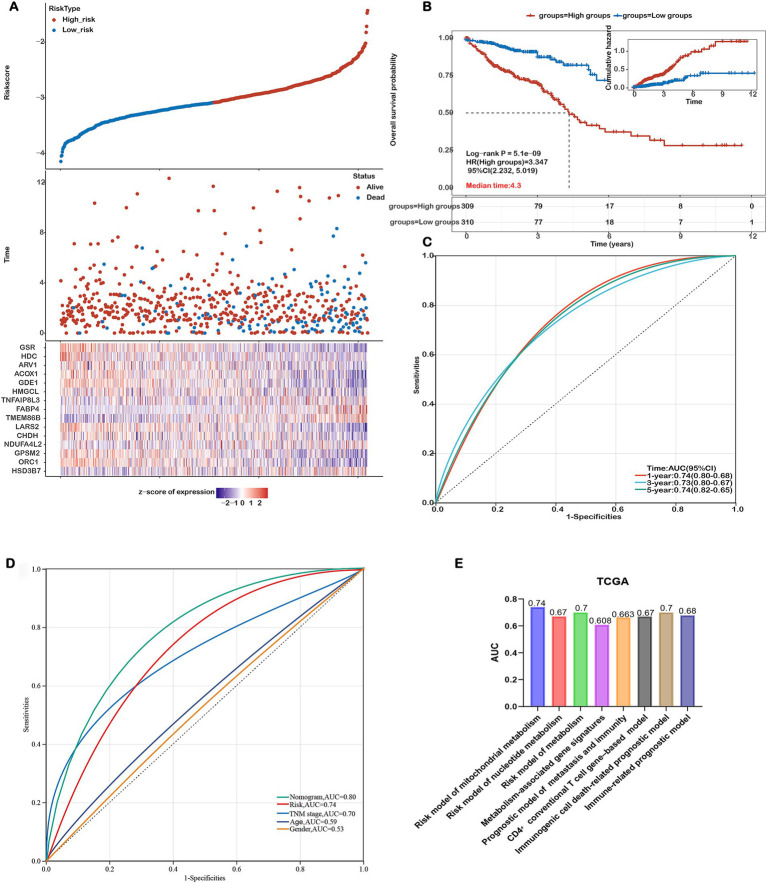
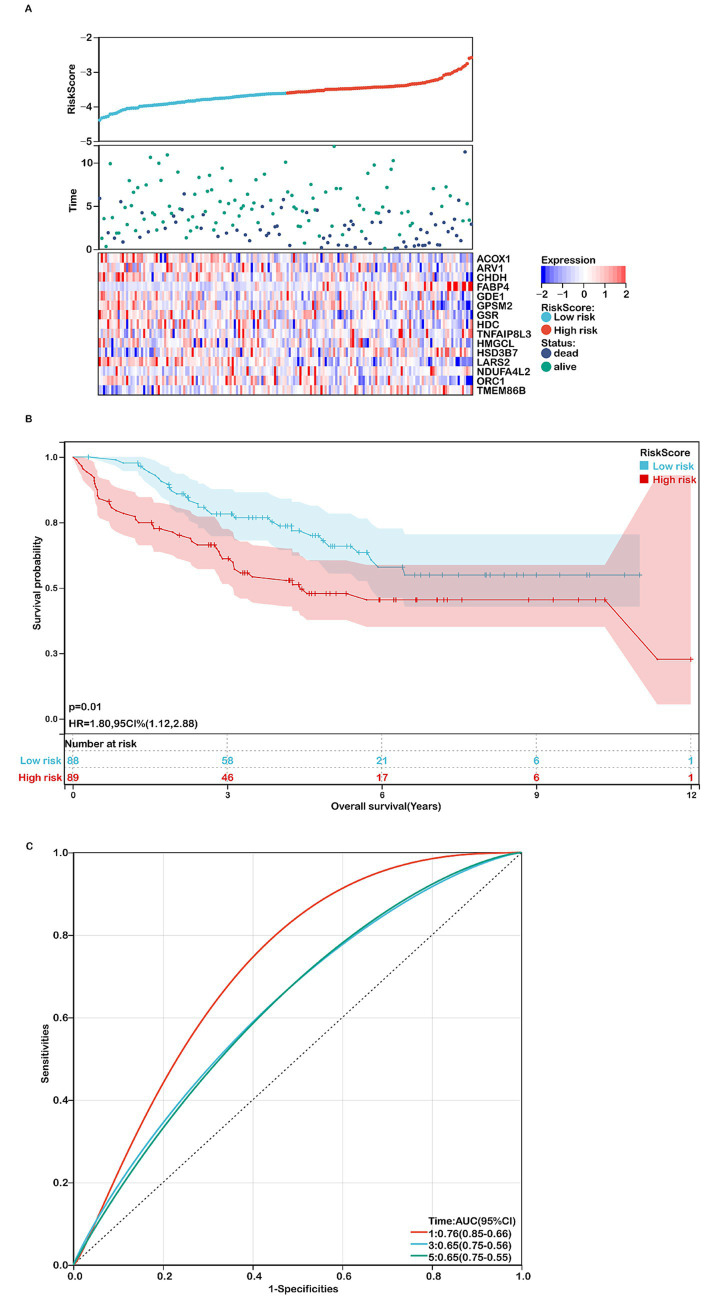
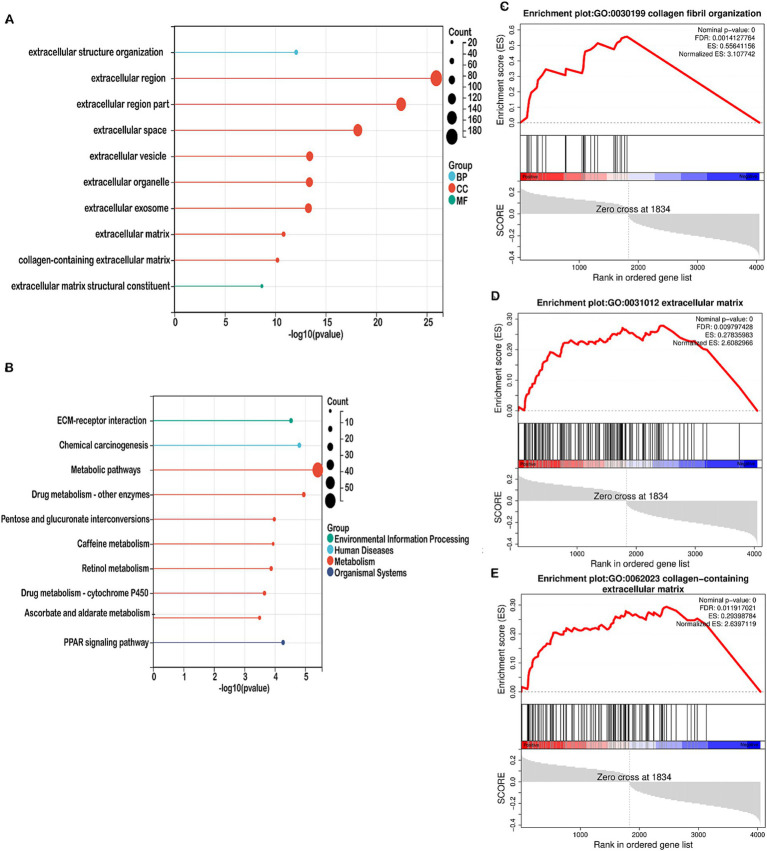
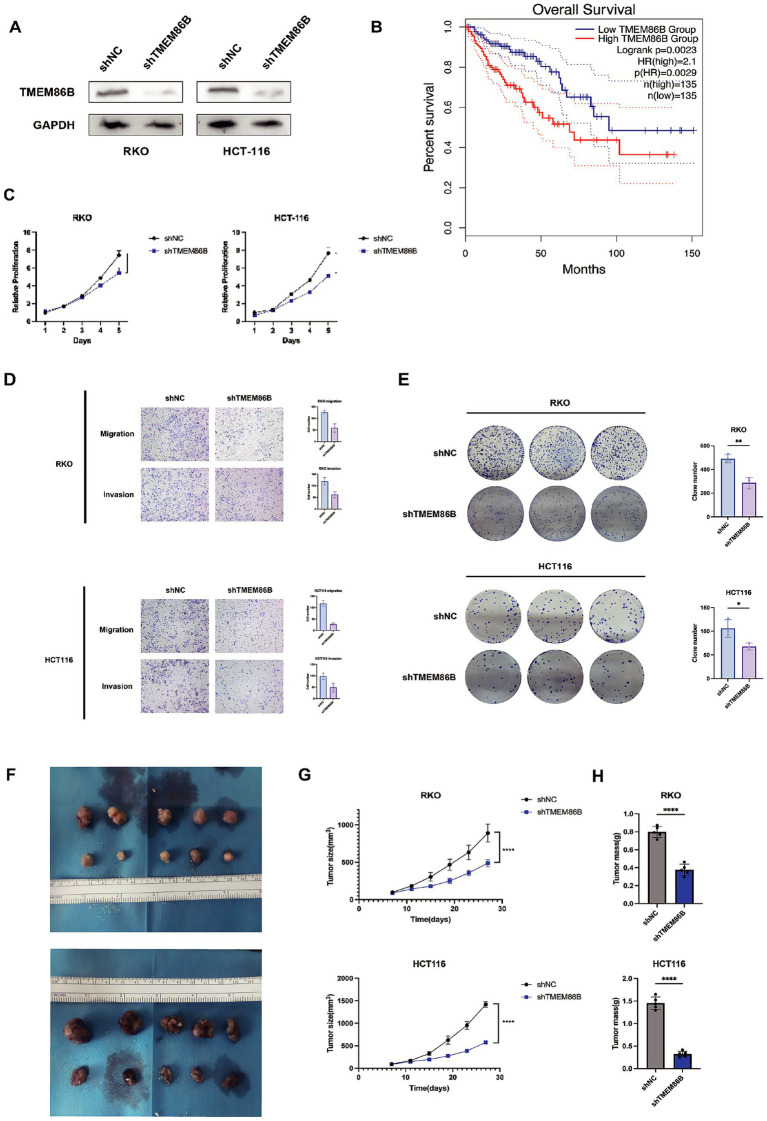
Methods: This study systematically analyzed the expression and prognostic value of mitochondrial metabolism-related genes in CRC patients, establishing a risk model using data from TCGA and GEO databases. We also investigated the tumor microenvironment (TME), immune cell infiltration, tumor mutation burden, microsatellite instability (MSI), and drug sensitivity. Core mitochondrial metabolism-related gene, TMEM86B was identified and its functions validated through cell-based assays and in vivo mouse models.
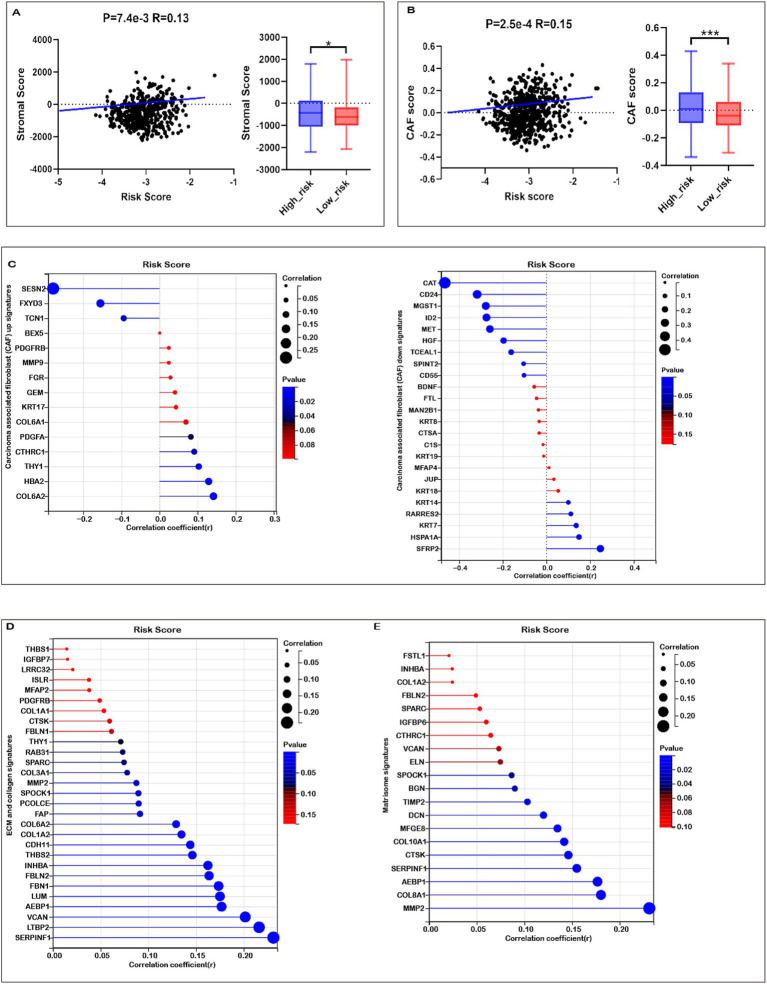
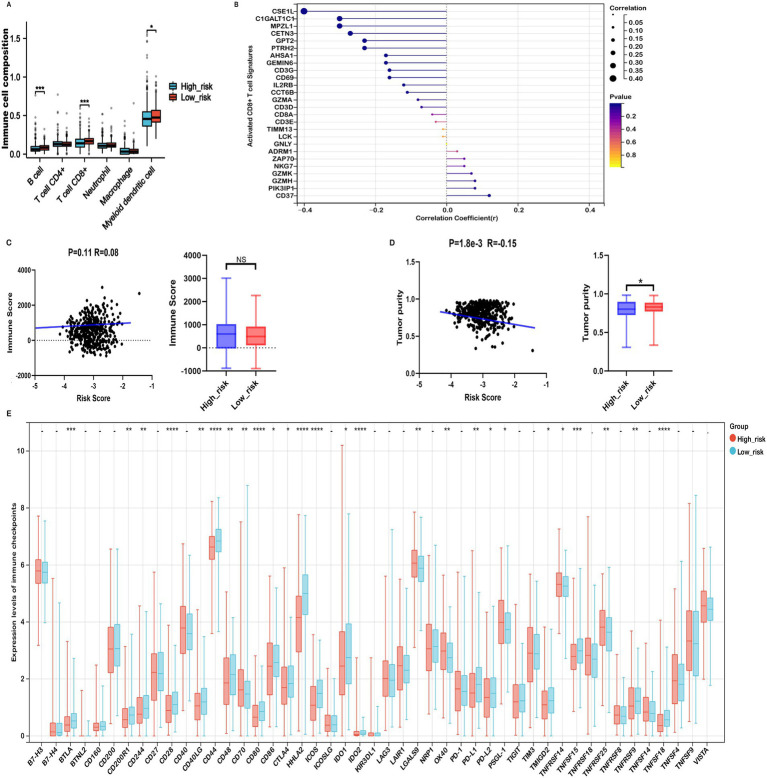
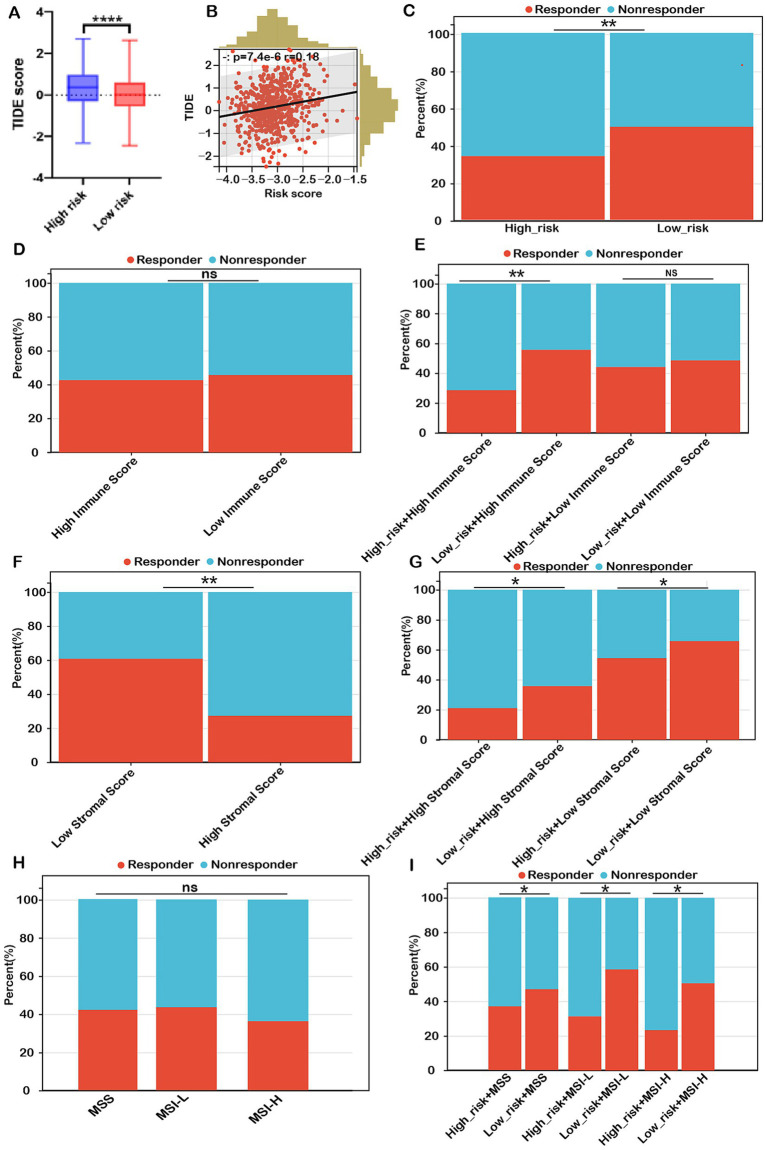
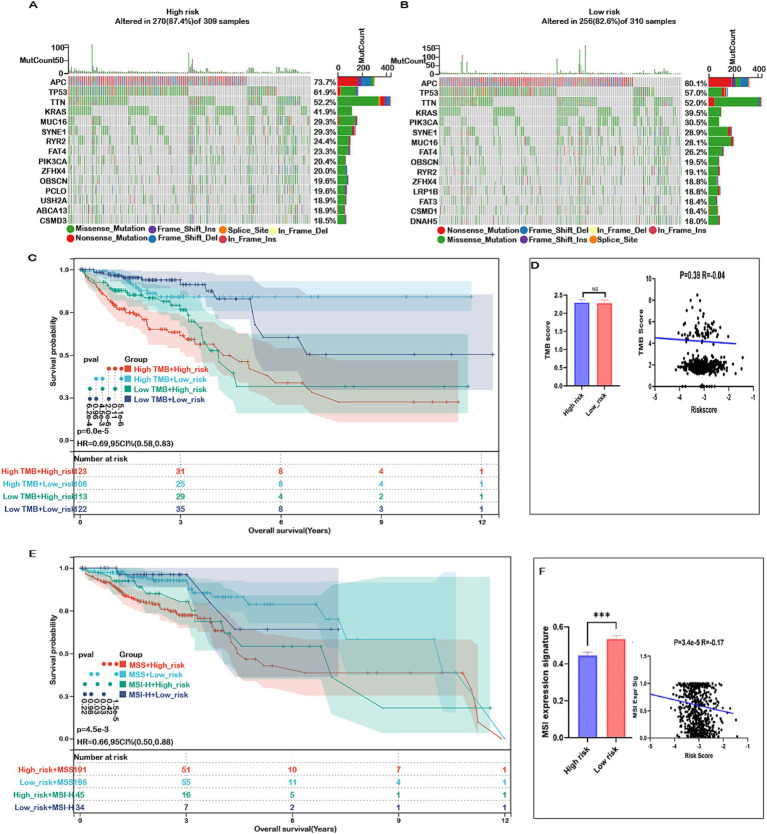
Results: Fifteen mitochondrial metabolism-related genes were identified, including HSD3B7, ORC1, GPSM2, NDUFA4L2, CHDH, LARS2, TMEM86B, FABP4, TNFAIP8L3, HMGCL, GDE1, ACOX1, ARV1, HDC, and GSR. The nomogram, which incorporates independent prognostic genes TMEM86B, TNFAIP8L3, HDC, and key clinical features pTNM stage (pathological Tumor-Node-Metastasis), age, was created to predict patient outcomes. Notable differences in immune cell infiltration were observed between risk groups. The risk score was associated with TME genes and immune checkpoints, indicating an immunosuppressive environment in high-risk groups. Furthermore, TIDE analysis revealed that integrating the risk score with immune score, stromal score, or microsatellite status improved the prediction of immunotherapy response across different CRC patient subgroups. Core mitochondrial metabolism-related gene, TMEM86B promotes colorectal cancer progression by enhancing cell proliferation, migration, and invasion, and its downregulation significantly inhibits tumor growth both in vitro and in vivo.
Conclusion: Our findings indicate that the risk model associated with mitochondrial metabolism may serve as a dependable prognostic indicator, facilitating tailored therapeutic strategies for CRC patients. TMEM86B promotes colorectal cancer progression, and its downregulation inhibits tumor growth in vitro and in vivo.
Keywords: colorectal cancer; drug susceptibility; immunotherapy; mitochondrial metabolism; prognostic biomarker; tumor microenvironment.
Copyright © 2025 Wang, Zhang and Ning.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures


Similar articles
-
Construction and validation of a lipid metabolism-related genes prognostic signature for skin cutaneous melanoma.Biochem Biophys Res Commun. 2025 Aug 15;775:152115. doi: 10.1016/j.bbrc.2025.152115. Epub 2025 May 29. Biochem Biophys Res Commun. 2025. PMID: 40460484
-
Comprehensive pan-cancer analysis reveals NTN1 as an immune infiltrate risk factor and its potential prognostic value in SKCM.Sci Rep. 2025 Jan 25;15(1):3223. doi: 10.1038/s41598-025-85444-x. Sci Rep. 2025. PMID: 39863609 Free PMC article.
-
Identification and validation of a KRAS-macrophage-associated gene signature as prognostic biomarkers and potential therapeutic targets in melanoma.Front Immunol. 2025 Jun 18;16:1566432. doi: 10.3389/fimmu.2025.1566432. eCollection 2025. Front Immunol. 2025. PMID: 40607411 Free PMC article.
-
Cost-effectiveness of using prognostic information to select women with breast cancer for adjuvant systemic therapy.Health Technol Assess. 2006 Sep;10(34):iii-iv, ix-xi, 1-204. doi: 10.3310/hta10340. Health Technol Assess. 2006. PMID: 16959170
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
References
LinkOut - more resources
Full Text Sources
Research Materials