

Chk2 deletion rescues bone loss and cellular senescence induced by Bmi1 deficiency via regulation of Cyp1a1
- PMID: 40698069
- PMCID: PMC12282458
- DOI: 10.1016/j.jot.2025.04.014
Chk2 deletion rescues bone loss and cellular senescence induced by Bmi1 deficiency via regulation of Cyp1a1
Erratum in
-
Corrigendum to "Chk2 deletion rescues bone loss and cellular senescence induced by Bmi1 deficiency via regulation of Cyp1a1" [J Orthop Translat, 52 (2025) 360-375 /doi.org/10.1016/j.jot.2025.04.014].J Orthop Translat. 2025 Jun 4;53:37. doi: 10.1016/j.jot.2025.05.009. eCollection 2025 Jul. J Orthop Translat. 2025. PMID: 40525096 Free PMC article.
Abstract
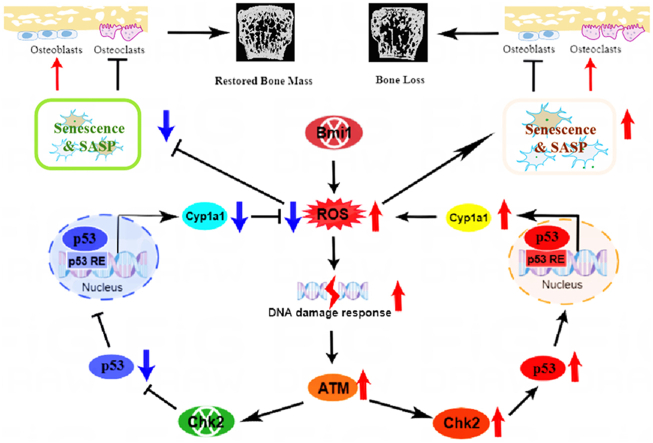
Background/objective: Bone homeostasis, maintained by a balance between osteoblastic bone formation and osteoclastic bone resorption, is disrupted in osteoporosis, leading to reduced bone mass and increased fracture risk. Bmi1, a polycomb group protein, is crucial for stem cell self-renewal and senescence regulation. Bmi1 deficiency has been linked to oxidative stress, DNA damage, and premature osteoporosis. Checkpoint kinase 2 (Chk2) is a key mediator of the DNA damage response (DDR) pathway, which can exacerbate bone aging through oxidative stress and senescence. This study investigated the role of Chk2 deletion in mitigating bone loss and cellular senescence caused by Bmi1 deficiency and explored the underlying molecular mechanisms, focusing on the regulation of oxidative stress via Cyp1a1.
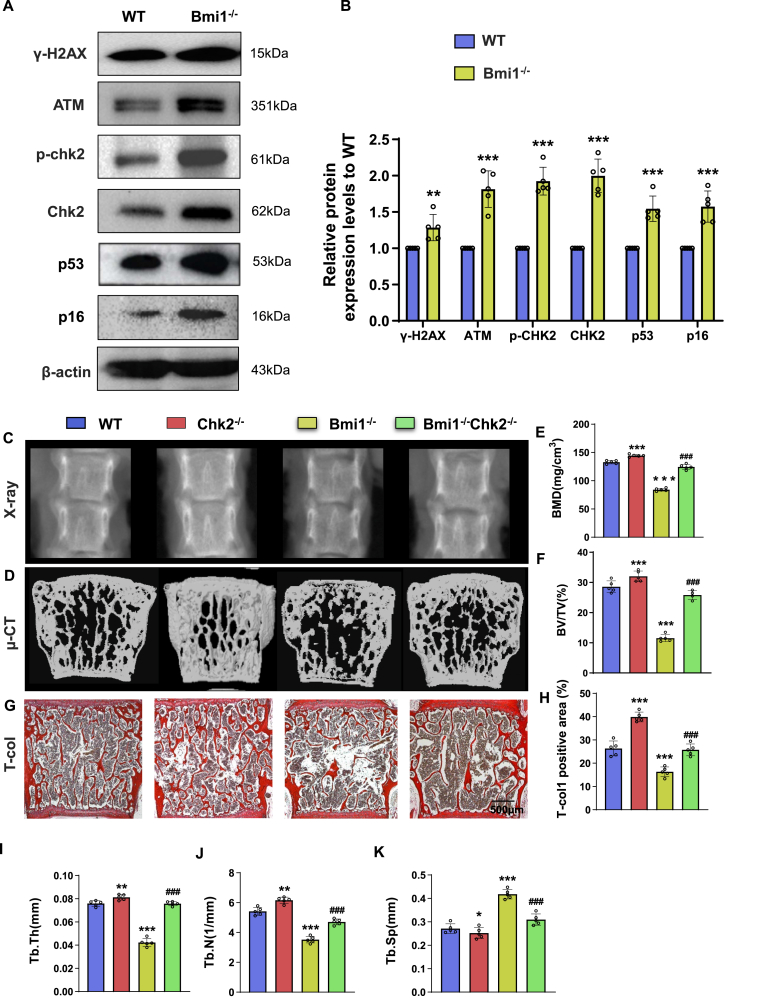
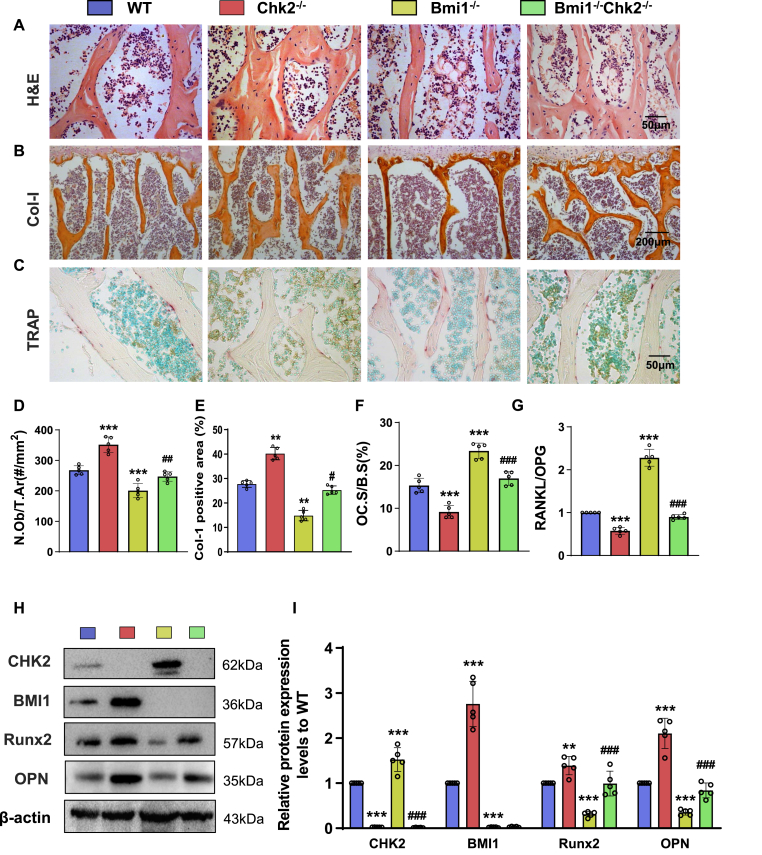
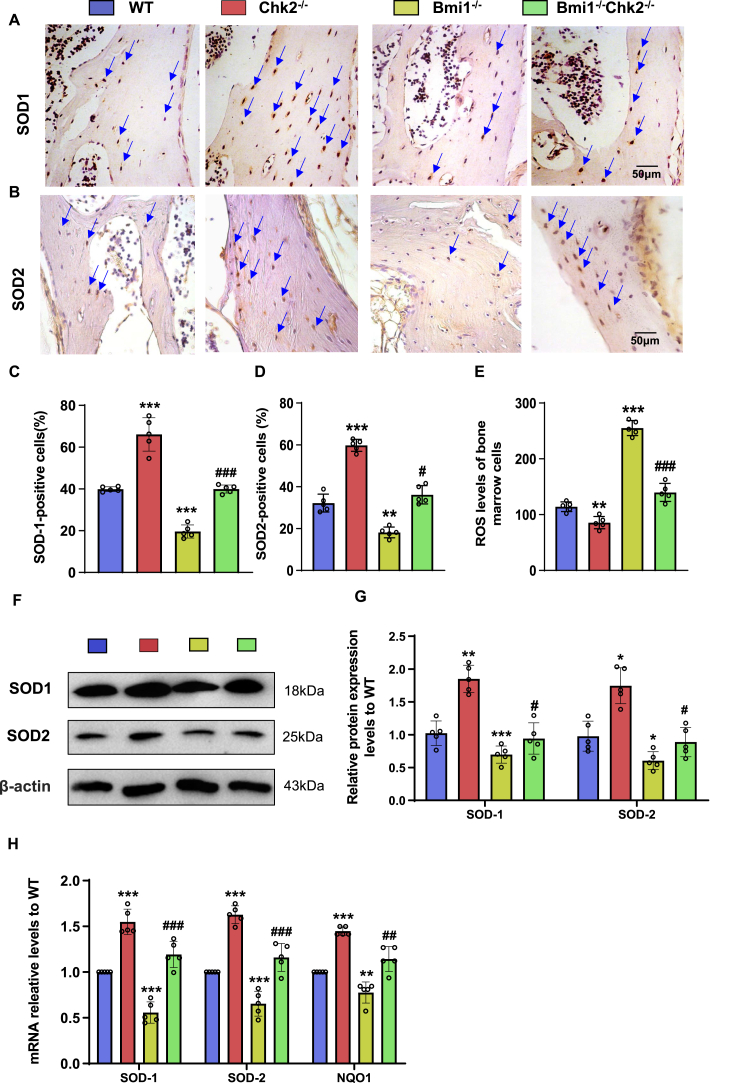
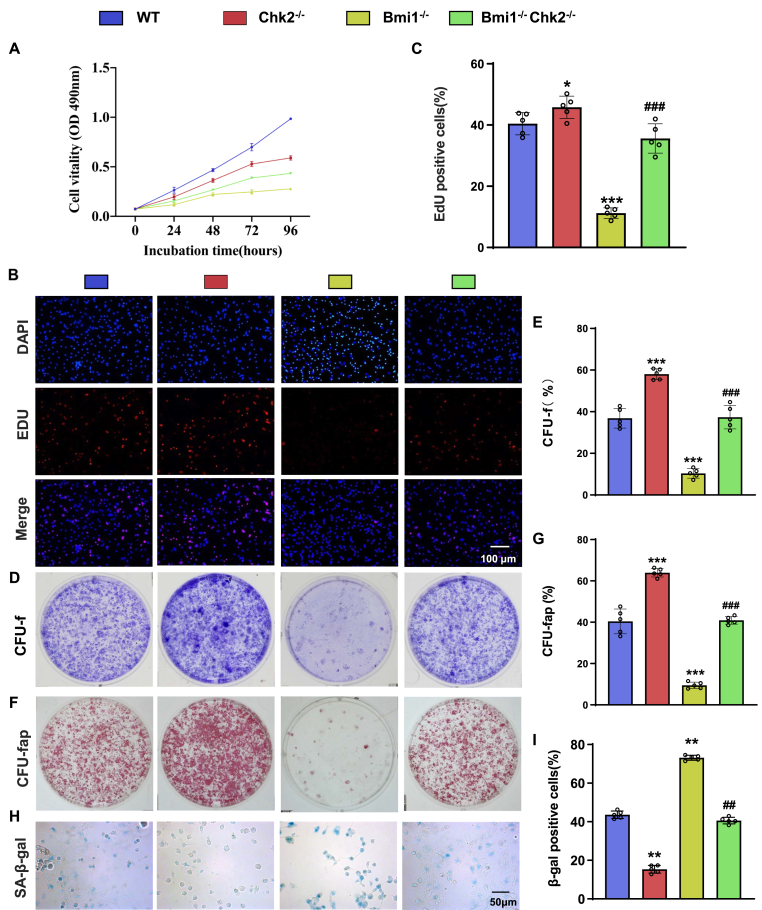
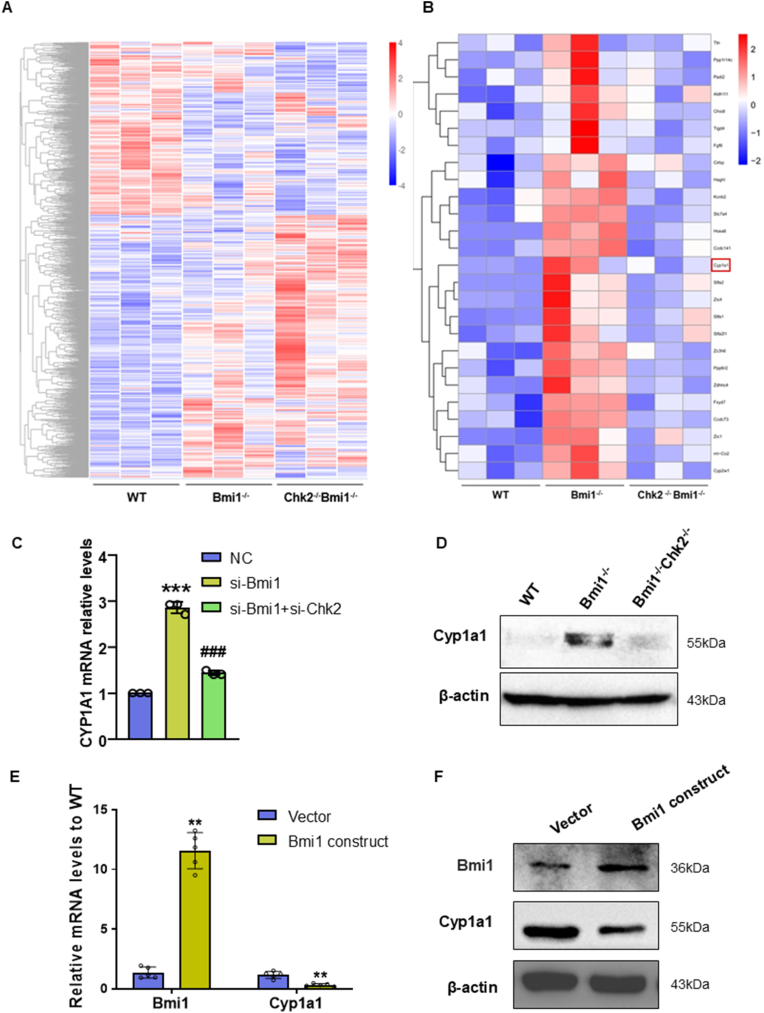
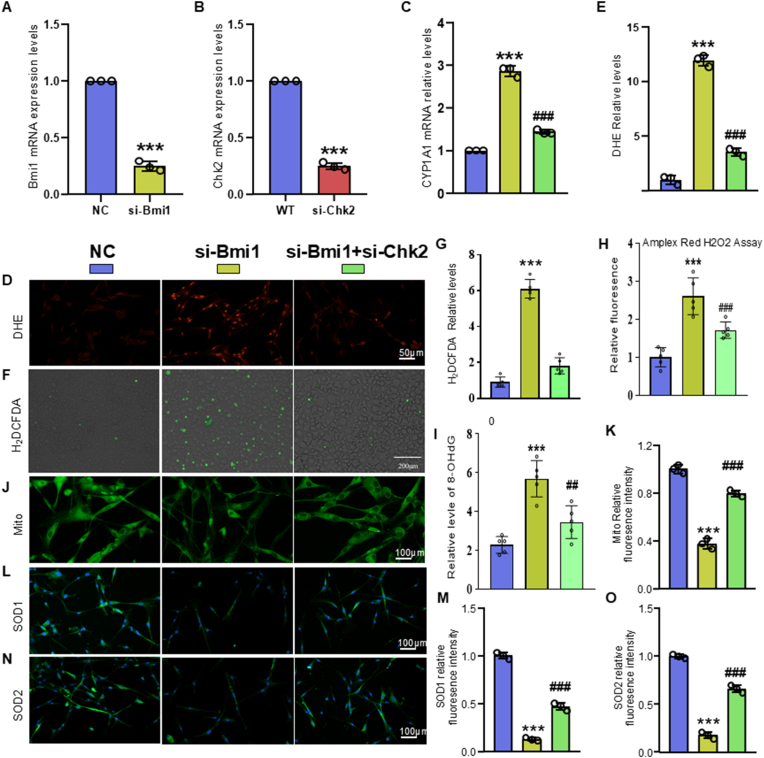
Methods: We utilized Bmi1-deficient (Bmi1-/-), Chk2-deficient (Chk2-/-), and double knockout (Bmi1-/-Chk2-/-) mice to assess bone homeostasis. Bone mineral density (BMD), trabecular architecture, and bone turnover markers were evaluated using X-ray imaging, micro-CT, histological staining, and bone histomorphometry. Oxidative stress markers, DDR pathway activation, and senescence-associated secretory phenotype (SASP) were analyzed using Western blotting, immunohistochemistry, and real-time PCR. Transcriptome sequencing identified differentially expressed genes, including Cyp1a1, which was further validated through chromatin immunoprecipitation (ChIP), luciferase assays, and knockdown experiments in bone marrow mesenchymal stem cells (BMSCs).
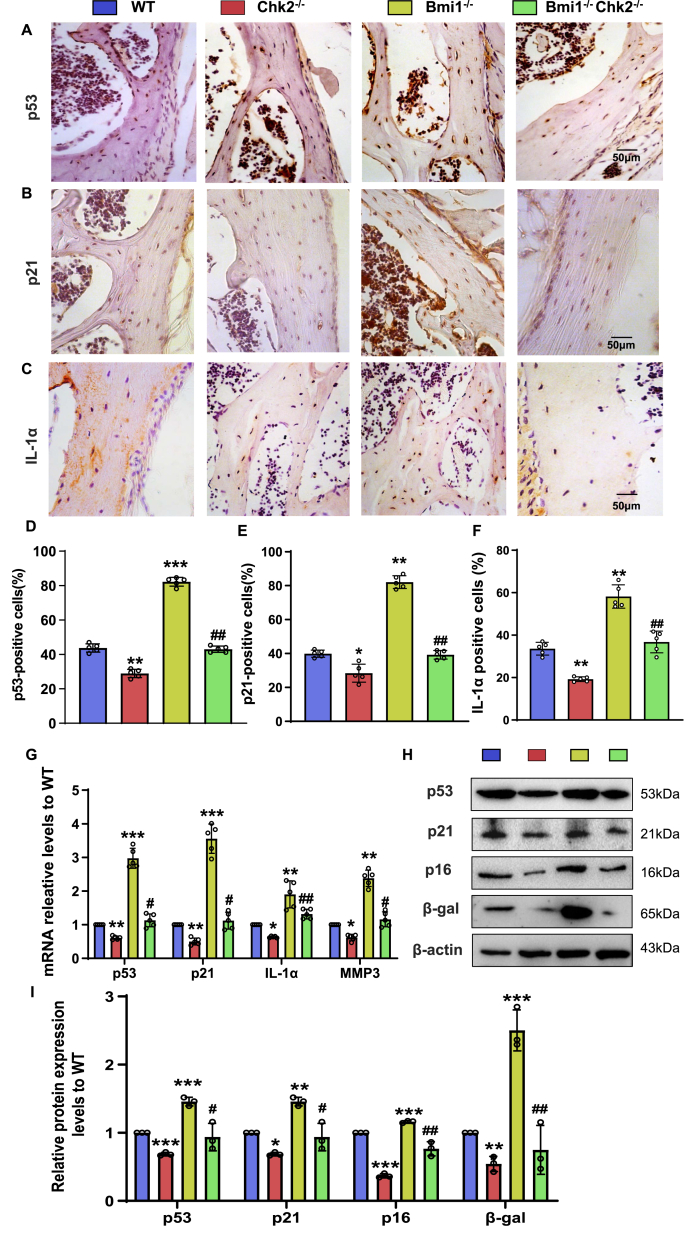
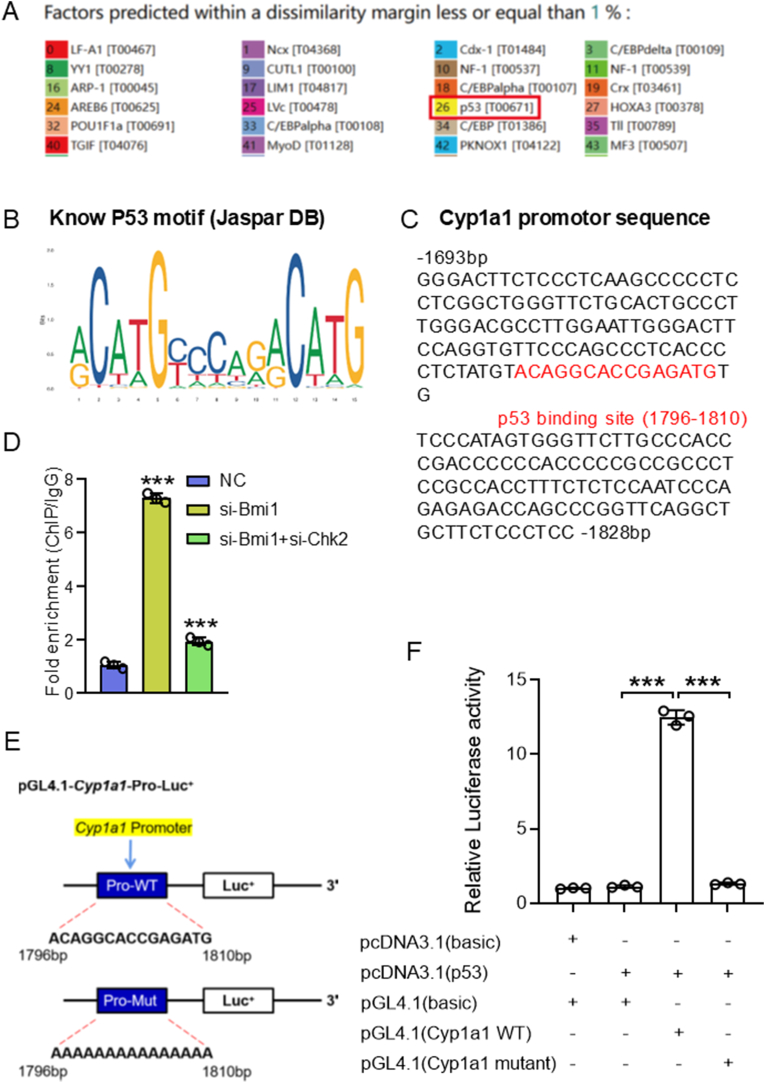
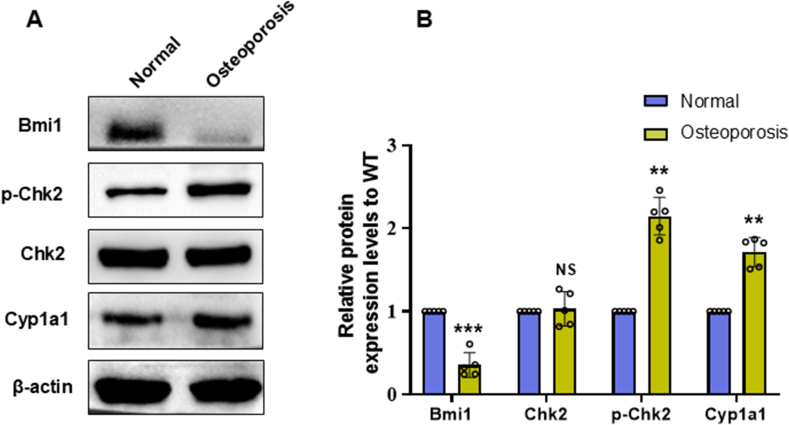
Results: Bmi1 deficiency activated the ATM-Chk2-p53 DDR pathway, increased oxidative stress, and induced osteocyte senescence and senescence-associated secretory phenotype (SASP), leading to reduced osteoblastic bone formation, increased osteoclastic bone resorption, and significant bone loss. Chk2 knockout rescued these defects by reducing oxidative stress and senescence. In Bmi1-/-Chk2-/- mice, BMD, trabecular bone volume, collagen deposition, and osteoblast markers (Runx2 and OPN) were significantly improved, while osteoclast markers (TRAP and RANKL/OPG ratio) were reduced compared to Bmi1-/- mice. Oxidative stress markers, including SOD1 and SOD2, were restored, and senescence markers such as p16, p21, and β-gal activity were significantly decreased. Transcriptome analysis identified Cyp1a1 as a key regulator of oxidative stress downstream of Bmi1 and Chk2. Bmi1 deficiency upregulated Cyp1a1, increasing ROS levels, while Chk2 knockout downregulated Cyp1a1 and mitigated oxidative stress. Mechanistically, p53 was shown to directly bind the Cyp1a1 promoter and activate its transcription, with Chk2 knockout reducing p53-mediated Cyp1a1 expression. These findings highlight the critical role of the Bmi1-Chk2-p53-Cyp1a1 axis in regulating bone homeostasis.
Conclusion: Chk2 knockout rescues bone loss and cellular senescence induced by Bmi1 deficiency by reducing oxidative stress through downregulation of Cyp1a1. These findings provide novel insights into the molecular mechanisms underlying bone aging and identify Chk2 and Cyp1a1 as potential therapeutic targets for osteoporosis and age-related bone disorders.
The translational potential of this article: This study identifies Chk2 and Cyp1a1 as potential therapeutic targets for osteoporosis and age-related bone loss. Targeting Chk2 or Cyp1a1 could mitigate oxidative stress and cellular senescence, offering a novel approach to preserving bone mass and preventing fractures in aging populations.
Keywords: Bmi1; Cellular senescence; Chk2; Cyp1a1; Osteoporosis; Oxidative stress.
© 2025 The Authors.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13(7):791–801. - PubMed
-
- Park I.K., Qian D., Kiel M., Becker M.W., Pihalja M., Weissman I.L., et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–305. - PubMed
-
- Zhang H.W., Ding J., Jin J.L., Guo J., Liu J.N., Karaplis A., et al. Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J Bone Miner Res. 2010;25(3):640–652. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
