

Unraveling the role of STAT3 in Cancer Cachexia: pathogenic mechanisms and therapeutic opportunities
- PMID: 40704145
- PMCID: PMC12283293
- DOI: 10.3389/fendo.2025.1608612
Unraveling the role of STAT3 in Cancer Cachexia: pathogenic mechanisms and therapeutic opportunities
Abstract
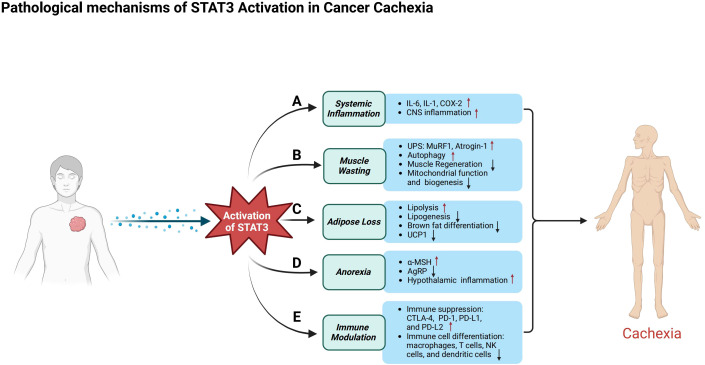
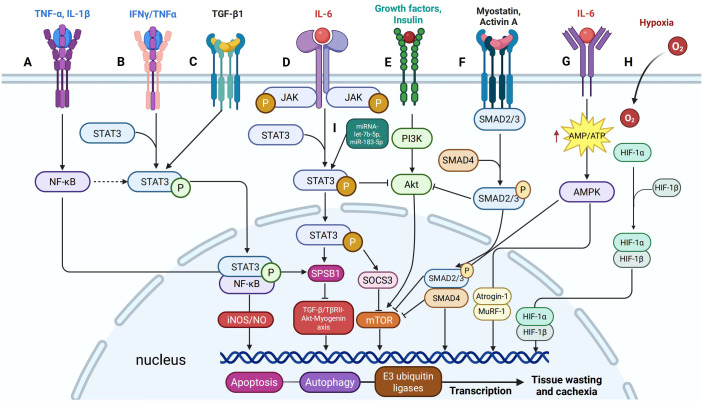
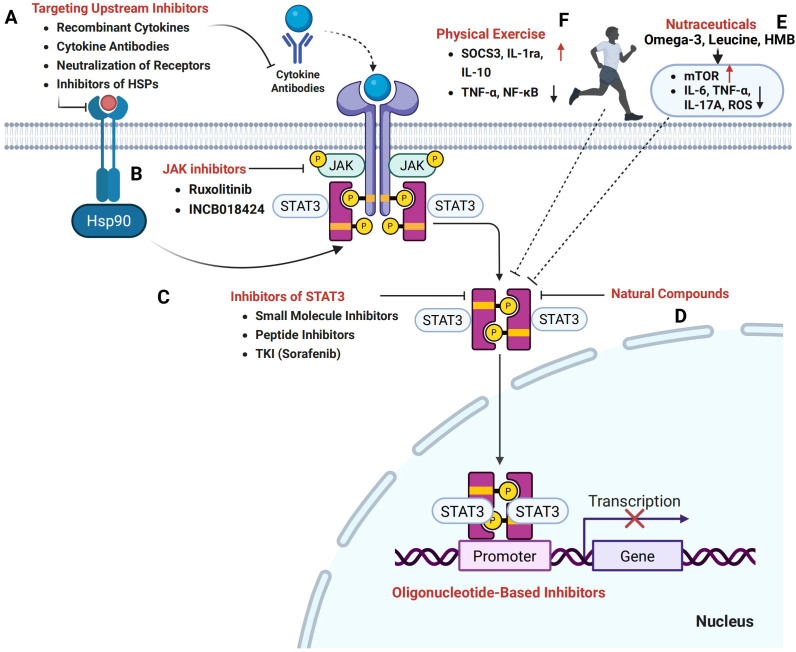
Cancer cachexia is a complex, multifactorial syndrome characterized by severe weight loss, muscle wasting, and systemic inflammation, significantly contributing to cancer-related morbidity and mortality. Signal transducer and activator of transcription 3 (STAT3) has emerged as a central mediator in the pathogenesis of this multifactorial condition. STAT3 regulates a broad range of cellular processes including inflammation, proteolysis, and mitochondrial dysfunction across multiple tissues, particularly skeletal muscle and adipose tissue. Persistent activation of STAT3 in response to tumor-derived and host-derived cytokines drives catabolic signaling cascades, disrupts anabolic pathways, and impairs energy homeostasis. Recent studies have illuminated the cross-talk between STAT3 and other signaling pathways that exacerbate cachexia-related metabolic imbalances. These findings position STAT3 not only as a critical mediator of cachexia progression but also as a promising therapeutic target. Pharmacological inhibition of STAT3 signaling has demonstrated efficacy in preclinical models, offering potential avenues for clinical intervention. This review provides a comprehensive overview of the molecular mechanisms by which STAT3 contributes to cancer cachexia and discusses emerging therapeutic strategies aimed at modulating STAT3 activity to mitigate the progression of this debilitating syndrome.
Keywords: STAT3; cancer cachexia; muscle wasting; pathogenic mechanisms; systemic inflammation; therapeutic strategies.
Copyright © 2025 Lv and Ding.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Cancer-Induced Muscle Wasting Requires p38β MAPK Activation of p300.Cancer Res. 2021 Feb 15;81(4):885-897. doi: 10.1158/0008-5472.CAN-19-3219. Epub 2020 Dec 22. Cancer Res. 2021. PMID: 33355181 Free PMC article.
-
The Power of Drosophila in Modeling Cancer Cachexia.Adv Exp Med Biol. 2025;1482:83-100. doi: 10.1007/978-3-031-97035-1_5. Adv Exp Med Biol. 2025. PMID: 40745137 Review.
-
Bu-zhong-yi-qi decoction regulates JNK/c-JUN signaling pathway to improve skeletal muscle atrophy caused by cancer cachexia.J Ethnopharmacol. 2025 Jul 24;351:120078. doi: 10.1016/j.jep.2025.120078. Epub 2025 Jun 1. J Ethnopharmacol. 2025. PMID: 40460921
-
Systemic Inflammatory Response Syndrome.2025 Jun 20. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jun 20. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31613449 Free Books & Documents.
-
Cancer therapy and cachexia.J Clin Invest. 2025 Aug 1;135(15):e191934. doi: 10.1172/JCI191934. eCollection 2025 Aug 1. J Clin Invest. 2025. PMID: 40759570 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous