

Inhibition of the Caveolin-1 pathway promotes apoptosis and overcomes pan-tyrosine kinase inhibitor resistance in hepatocellular carcinoma
- PMID: 40715090
- PMCID: PMC12297225
- DOI: 10.1038/s41419-025-07887-4
Inhibition of the Caveolin-1 pathway promotes apoptosis and overcomes pan-tyrosine kinase inhibitor resistance in hepatocellular carcinoma
Abstract
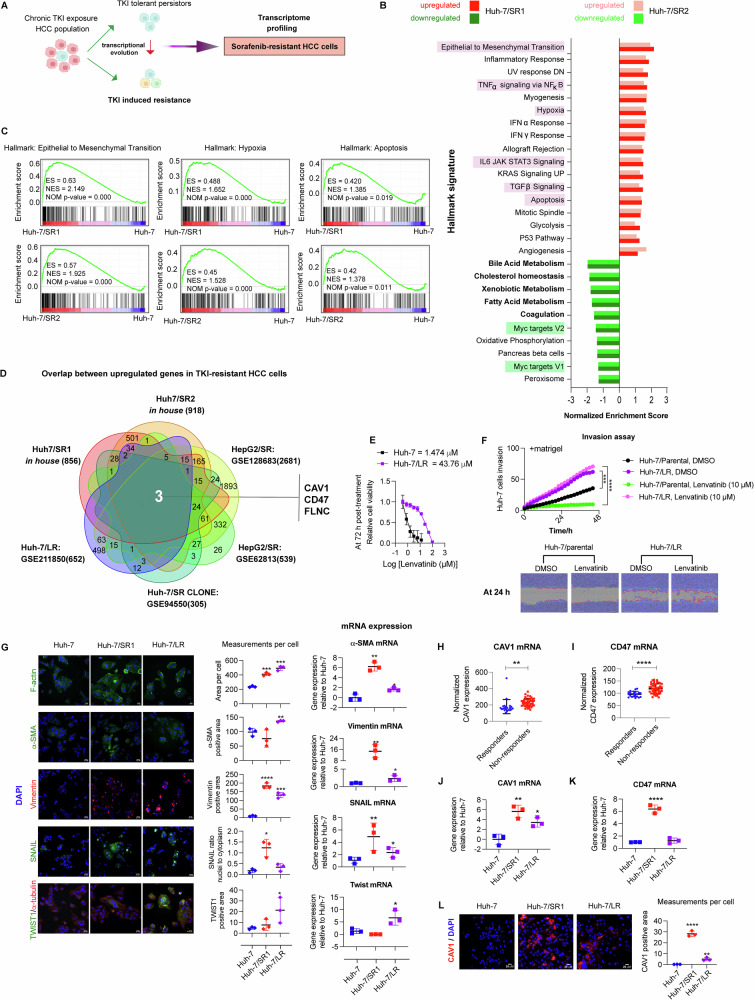
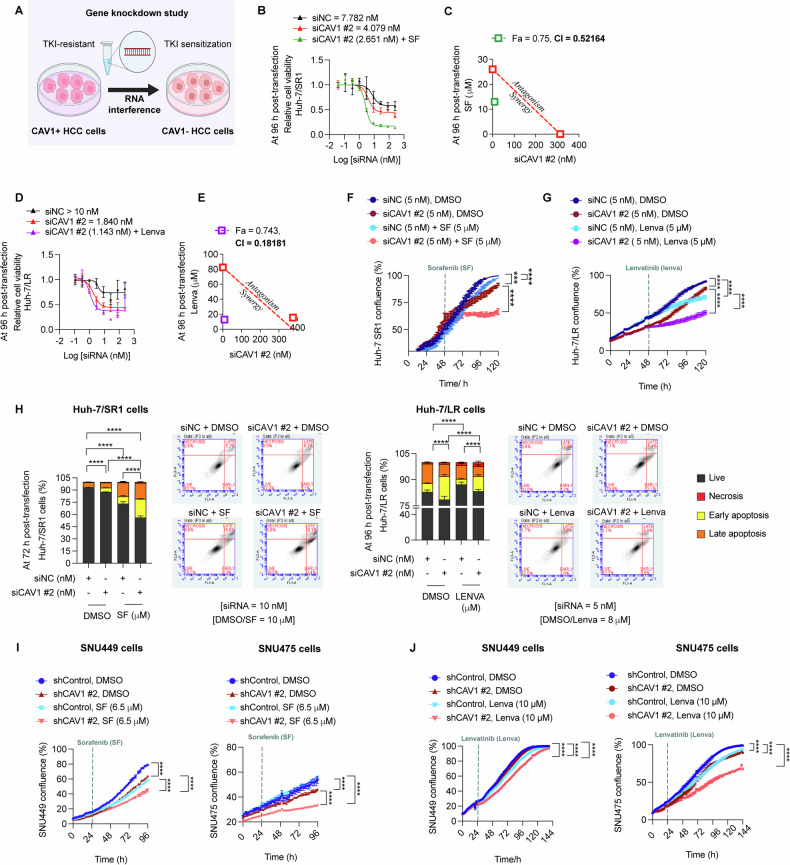
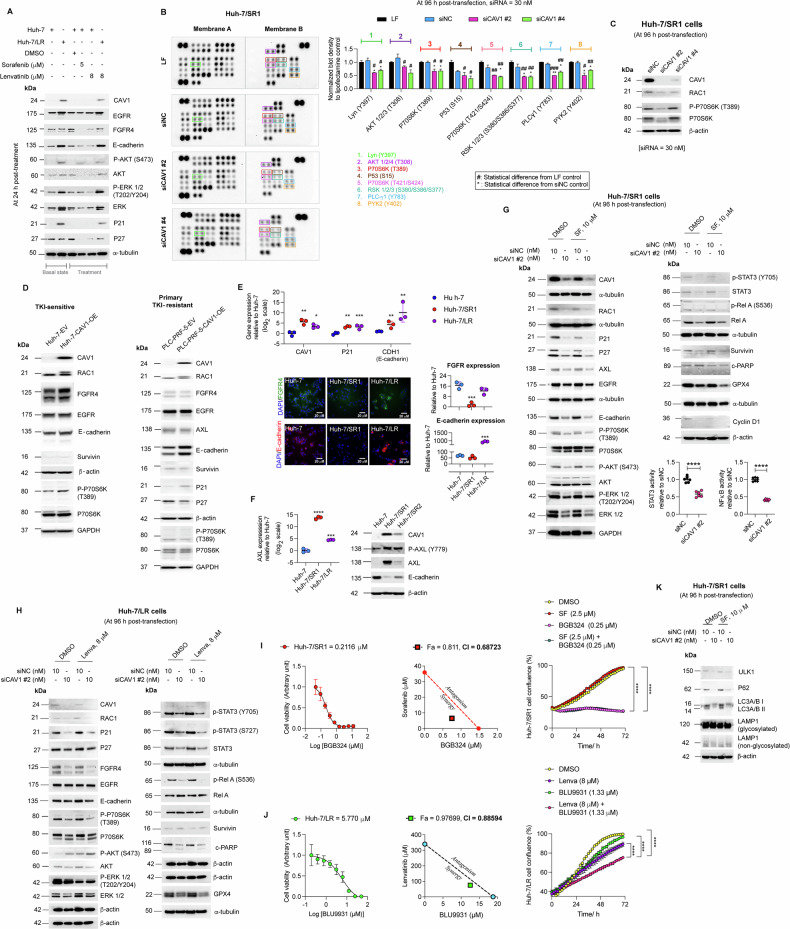
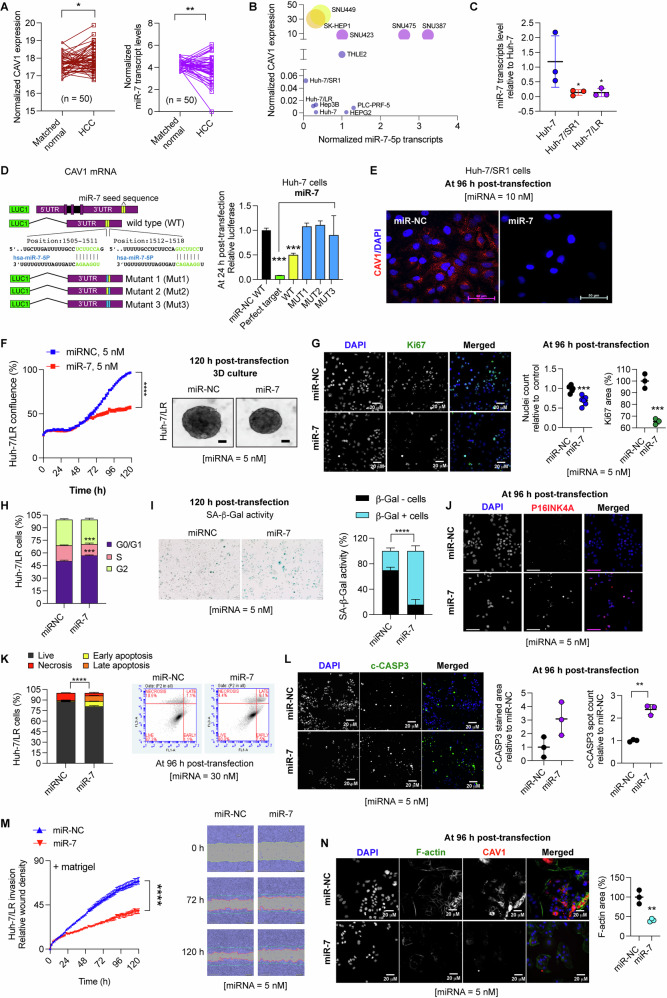
Resistance to multi-tyrosine kinase inhibitors (TKI) is a major clinical concern in advanced hepatocellular carcinoma (HCC). Herein, we aimed to uncover the mechanisms underlying pan-TKI resistance and to identify potential therapeutic targets. We used multiple TKI-resistant HCC cell lines to identify caveolin-1 (CAV1) as a key driver of therapeutic resistance. CAV1 downregulation induced apoptosis, inhibited metastasis and restored TKI sensitivity in both inherent and acquired TKI-resistant HCC cells. Mechanistically, in acquired TKI-resistant cells aberrant CAV1/STAT3/P70S6K signalling is required for their survival, motility, and invasiveness. CAV1 inhibition reduced expression of dormancy regulators E-cadherin, RAC1 and p21, enhanced cancer stemness markers, and disrupted downstream STAT3/P70S6K and AMPKα signalling pathways, prompting cancer cells to exit from dormancy and initiate autophagy-induced cell death. Furthermore, selective inhibition of AXL and FGFR4 downstream of the CAV1 pathway sensitized TKI-resistant cells to sorafenib and lenvatinib, respectively. In addition, microRNA-7-5p (miR-7) was identified as an endogenous regulator of CAV1; and miR-7's inhibitory effect on CAV1 and FGFR4 suppressed the STAT3/P70S6K pathway, promoted autophagy and triggered apoptosis in lenvatinib-resistant cells. Combination therapy using either lenvatinib or sorafenib and selective CAV1 inhibitors (e.g., siCAV1/miR-7), or AXL/FGFR4 inhibitors (e.g., BGB324/BLU9931) effectively overcame pan-TKI resistance. In HCC patient datasets, elevated CAV1 mRNA was observed in sorafenib non-responders, and single cell RNA-sequencing of HCC patient tumours revealed a rare population of CAV1+ cancer cells associated with recurrence. High CAV1 expression was specific to HBV+ HCC patients and independently predicted poor survival. Further, targeting of CAV1, AXL or FGFR4 effectively overcame TKI resistance in HCC patient derived organoids (PDOs). Our findings highlight a previously unrecognized role for CAV1-driven signalling in sustaining tumour dormancy, a critical and challenging therapeutic barrier underlying recurrence and pan-TKI resistance in HCC. Therapeutically targeting these pathways offer a promising and novel strategy to eliminate dormant tumour cells, thereby overcoming resistance and improving treatment outcomes.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethics approval and consent to participate: All methods were carried out in accordance with relevant guidelines and regulations and approved by the Sir Charles Gairdner Osborne Park Health Care Group Human Research Ethics Committee (approval number RGS0000000919). Informed consent was obtained from all participants, with anonymity and confidentiality maintained throughout the study. The research adhered to the ethical principles of the Declaration of Helsinki (2008) and was conducted in accordance with the Australian Code for the Responsible Conduct of Research (2018).
Figures




References
-
- Cheng Z, Wei-Qi J, Jin D. New insights on sorafenib resistance in liver cancer with correlation of individualized therapy. Biochim Biophys Acta Rev Cancer. 2020;1874:188382. - PubMed
-
- Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Prim. 2021;7:6. - PubMed
-
- Llovet JM, Kudo M, Merle P, Meyer T, Qin S, Ikeda M, et al. Lenvatinib plus pembrolizumab versus lenvatinib plus placebo for advanced hepatocellular carcinoma (LEAP-002): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2023;24:1399–410. - PubMed
-
- Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382:1894–905. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
