

Podocyte dysfunction driven by heme in sickle-cell nephropathy
- PMID: 40715197
- PMCID: PMC12297694
- DOI: 10.1038/s41598-025-09220-7
Podocyte dysfunction driven by heme in sickle-cell nephropathy
Abstract
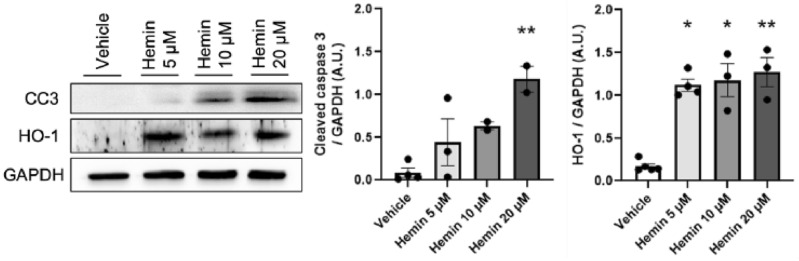
Sickle-cell disease (SCD) is characterized by vaso-occlusive crises and chronic hemolytic anemia, leading to tissue damage affecting various organs, including the kidneys. Hemolysis contributes to sickle-cell nephropathy (SCN) but the molecular mechanisms underlying the intravascular hemolysis and heme release involved in podocyte damage leading to proteinuria and chronic kidney disease remain uncertain. This study explored the impact of heme on podocyte function by exposing human podocytes cell line to hemin (5 μM hemin for 4 and 24 h), with or without the antioxidant N-acetyl cysteine (NAC). We then assessed the relevance of in vitro studies on renal biopsy specimens from controls with primary and secondary forms of focal segmental glomerulosclerosis (FSGS) and patients with SCD-related FSGS. After 4 h of hemin exposure, podocyte cytoskeleton alterations and increased apoptosis were observed. At 24 h, heme oxygenase-1 (HO-1) expression increased, alongside oxidative stress, DNA damage, and mitochondrial and endoplasmic reticulum dysfunctions. NF-κB pathway activation suggested an adaptive response. NAC partially reduced these effects, indicating oxidative stress's central role while implicating additional mechanisms in apoptosis induction. Renal biopsies from patients with focal segmental glomerulosclerosis (FSGS), including SCD-related cases, showed elevated HO-1 and BiP in podocytes compared to normal glomeruli, along with reduced synaptopodin, indicating damage. In conclusion, this study highlights the molecular mechanisms underlying heme-induced podocyte damage in SCN. Oxidative stress appears to play a key role, but other pathological pathways are also involved. These results open up new perspectives for understanding and treating SCN.
Keywords: Cellular stress; Heme; Hemolysis; Oxidative stress; Podocyte.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures










References
-
- Kato, G. J. et al. Sickle cell disease. Nat. Rev. Dis. Primers4, 18010. 10.1038/nrdp.2018.10 (2018). - PubMed
-
- Guasch, A., Navarrete, J., Nass, K. & Zayas, C. F. Glomerular involvement in adults with sickle cell hemoglobinopathies: prevalence and clinical correlates of progressive renal failure. J. Am. Soc. Nephrol.17, 2228–2235. 10.1681/ASN.2002010084 (2006). - PubMed
-
- Gérardin, C. et al. Survival and specific outcome of sickle cell disease patients after renal transplantation. Br. J. Haematol.187, 676–680. 10.1111/bjh.16113 (2019). - PubMed
-
- Nielsen, L. et al. Morbidity and mortality of sickle cell disease patients starting intermittent haemodialysis: a comparative cohort study with non- Sickle dialysis patients. Br. J. Haematol.10.1111/bjh.14040 (2016). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
