

Deep-learning structure elucidation from single-mutant deep mutational scanning
- PMID: 40715235
- PMCID: PMC12297490
- DOI: 10.1038/s41467-025-62261-4
Deep-learning structure elucidation from single-mutant deep mutational scanning
Abstract
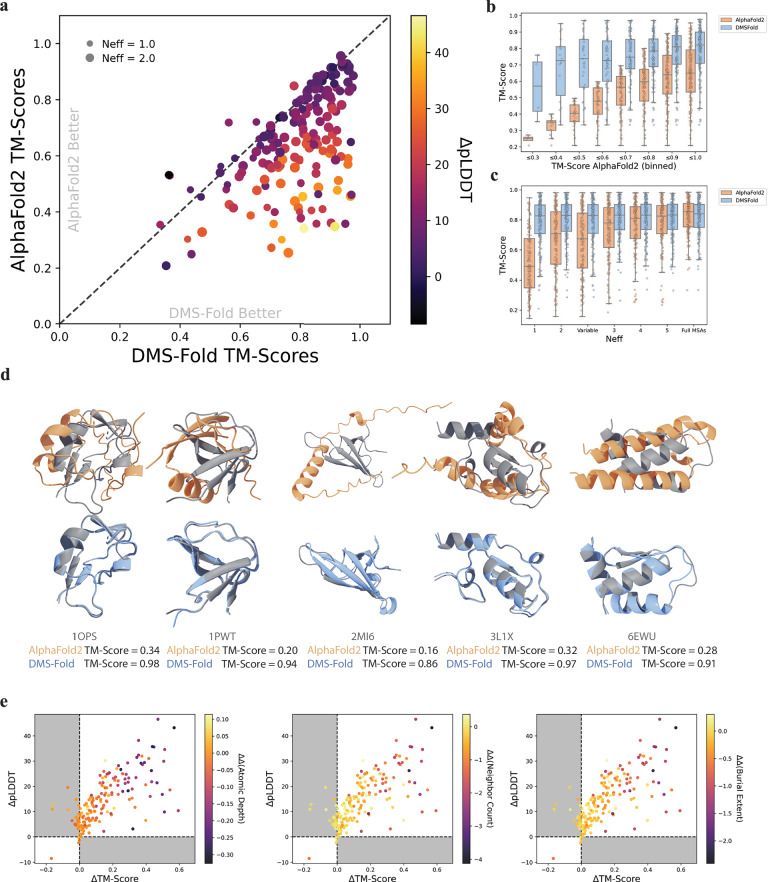
Deep learning has revolutionized the field of protein structure prediction. AlphaFold2, a deep neural network, vastly outperformed previous algorithms to provide near atomic-level accuracy when predicting protein structures. Despite its success, there still are limitations which prevent accurate predictions for numerous protein systems. Here we show that sparse residue burial restraints from deep mutational scanning (DMS) can refine AlphaFold2 to significantly enhance results. Burial information extracted from DMS is used to explicitly guide residue placement during structure generation. DMS-Fold was validated on both simulated and experimental single-mutant DMS, with DMS-Fold outperforming AlphaFold2 for 88% of protein targets and with 252 proteins having an improvement greater than 0.1 in TM-Score. DMS-Fold is free and publicly available: [ https://github.com/LindertLab/DMS-Fold ].
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




References
-
- Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science379, 1123–1130 (2023). - PubMed
-
- Wu, R. et al. High-resolution de novo structure prediction from primary sequence. bioRxiv. 2022.2007.2021.500999 (2022). 10.1101/2022.07.21.500999
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
