

AI-driven multi-omics modeling of myalgic encephalomyelitis/chronic fatigue syndrome
- PMID: 40715814
- PMCID: PMC12416096
- DOI: 10.1038/s41591-025-03788-3
AI-driven multi-omics modeling of myalgic encephalomyelitis/chronic fatigue syndrome
Abstract
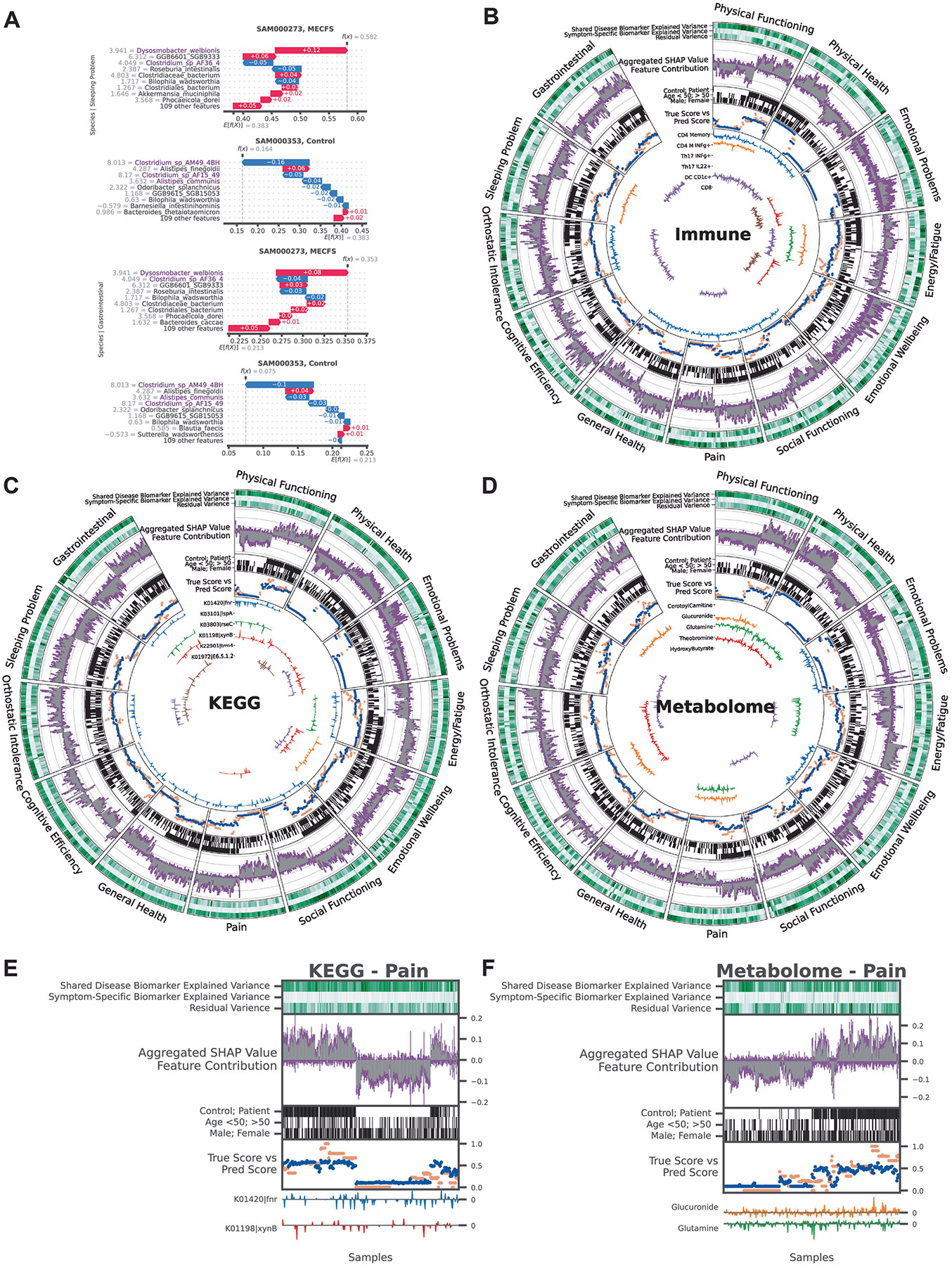
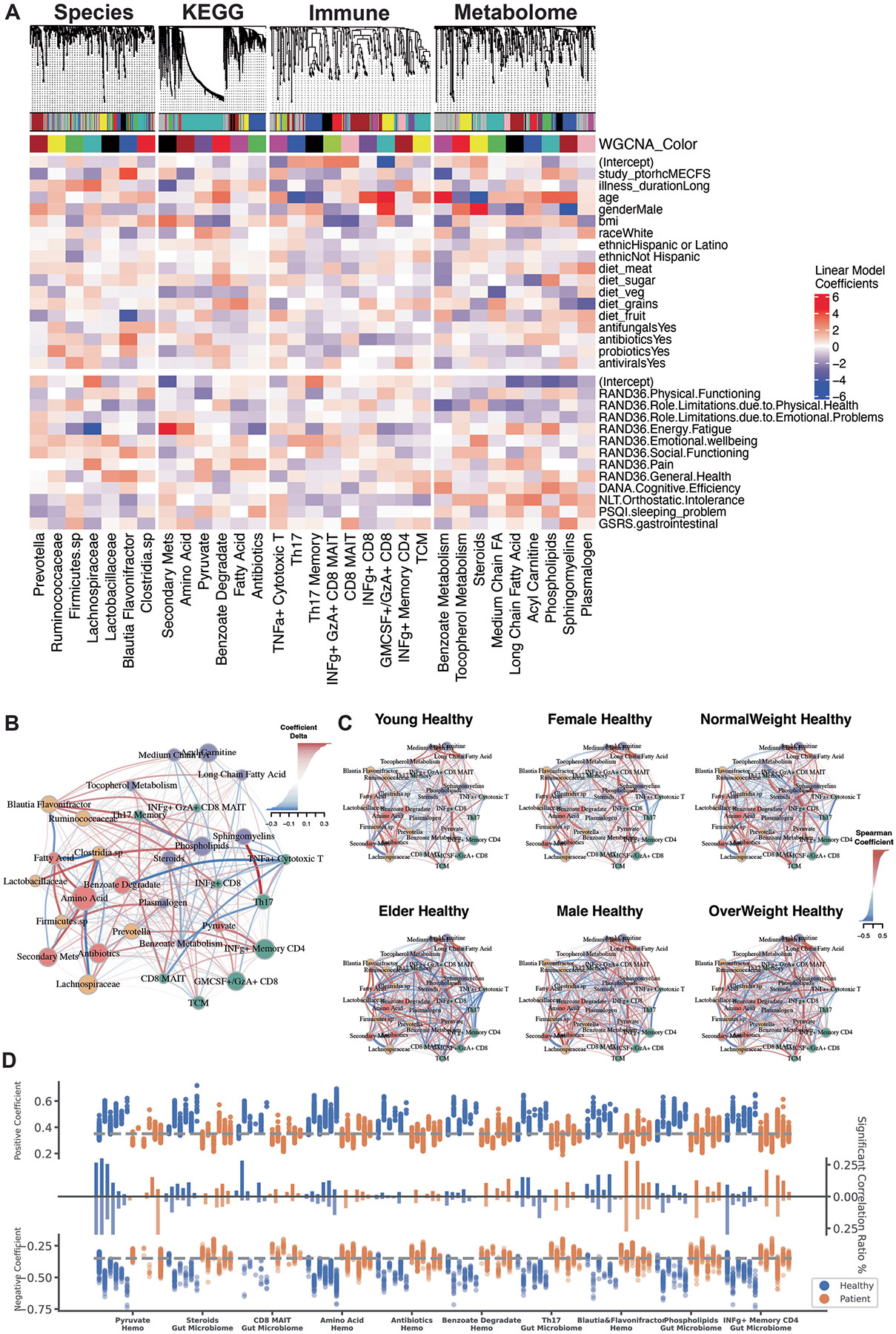
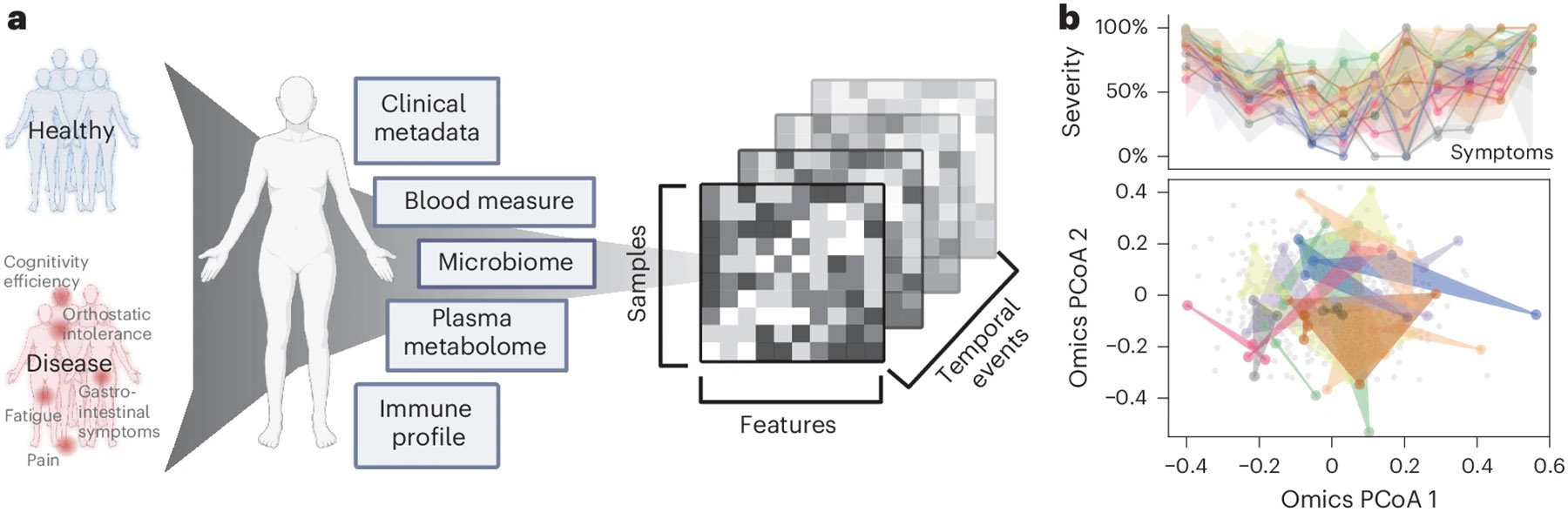
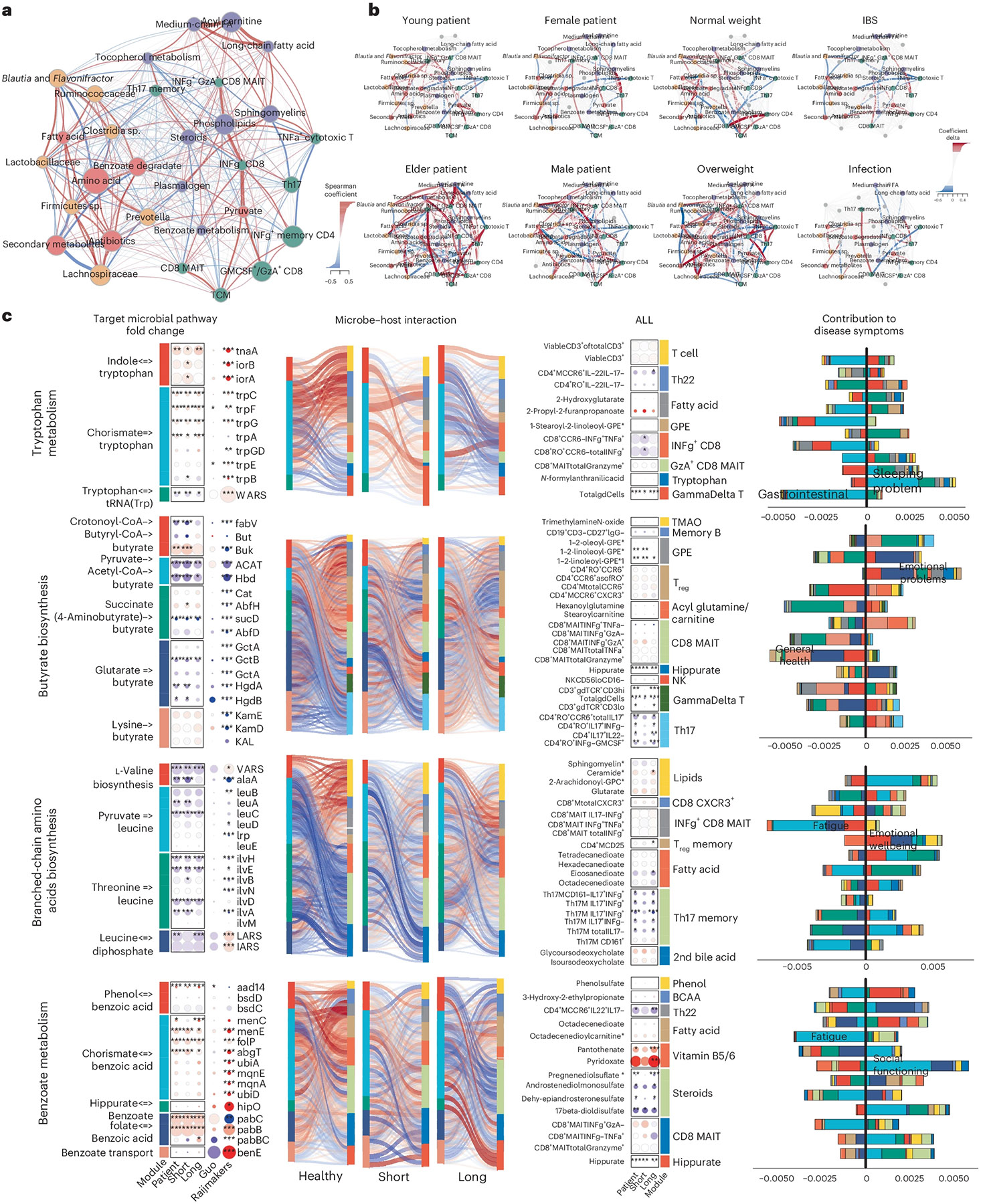
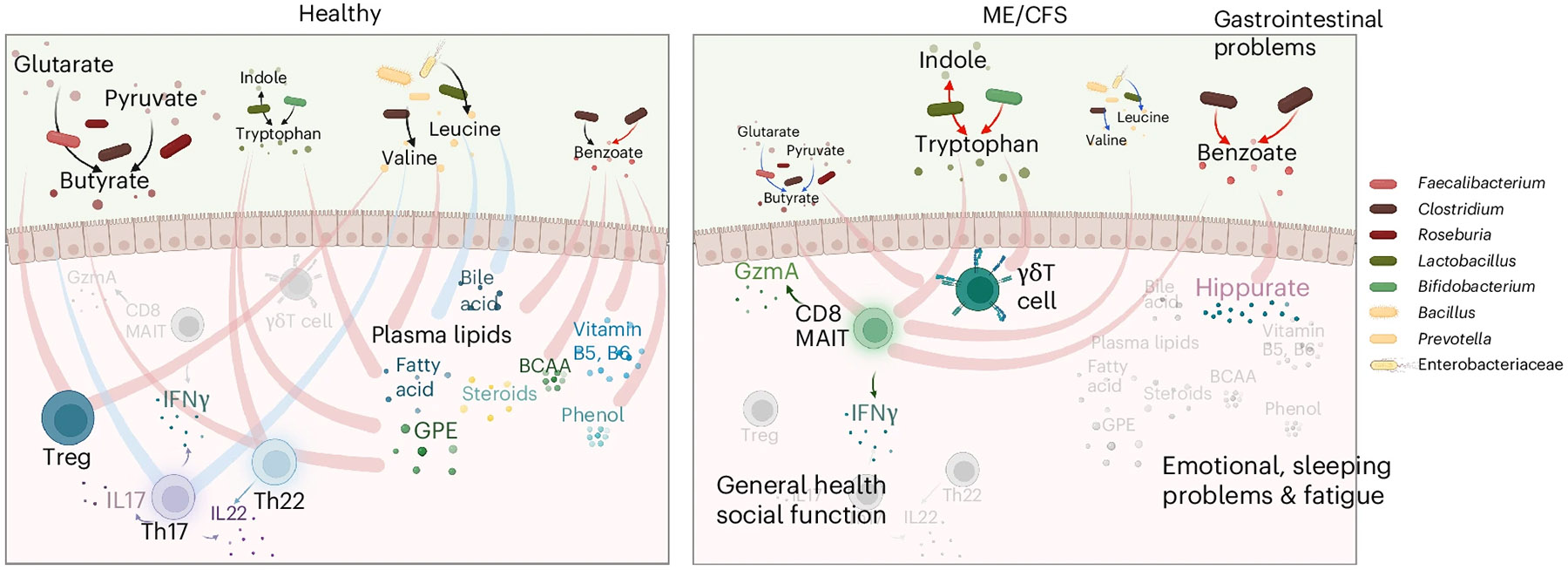
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a chronic illness with a multifactorial etiology and heterogeneous symptomatology, posing major challenges for diagnosis and treatment. Here we present BioMapAI, a supervised deep neural network trained on a 4-year, longitudinal, multi-omics dataset from 249 participants, which integrates gut metagenomics, plasma metabolomics, immune cell profiling, blood laboratory data and detailed clinical symptoms. By simultaneously modeling these diverse data types to predict clinical severity, BioMapAI identifies disease- and symptom-specific biomarkers and classifies ME/CFS in both held-out and independent external cohorts. Using an explainable AI approach, we construct a unique connectivity map spanning the microbiome, immune system and plasma metabolome in health and ME/CFS adjusted for age, gender and additional clinical factors. This map uncovers altered associations between microbial metabolism (for example, short-chain fatty acids, branched-chain amino acids, tryptophan, benzoate), plasma lipids and bile acids, and heightened inflammatory responses in mucosal and inflammatory T cell subsets (MAIT, γδT) secreting IFN-γ and GzA. Overall, BioMapAI provides unprecedented systems-level insights into ME/CFS, refining existing hypotheses and hypothesizing unique mechanisms-specifically, how multi-omics dynamics are associated to the disease's heterogeneous symptoms.
© 2025. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests: S.D.V. is affiliated and has a financial interest with The BioCollective, a company that provided the BioCollector, the collection kit used for at-home stool collection discussed in this paper. The other authors declare no competing interests.
Figures






References
-
- Cairns R & Hotopf M. A systematic review describing the prognosis of chronic fatigue syndrome. Occup. Med. Oxf. Engl 55, 20–31 (2005).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
