

Pushing the Thermodynamic and Kinetic Limits of Near-Infrared Emissive CrIII Complexes in Photocatalysis
- PMID: 40720722
- PMCID: PMC12333365
- DOI: 10.1021/jacs.5c08541
Pushing the Thermodynamic and Kinetic Limits of Near-Infrared Emissive CrIII Complexes in Photocatalysis
Abstract
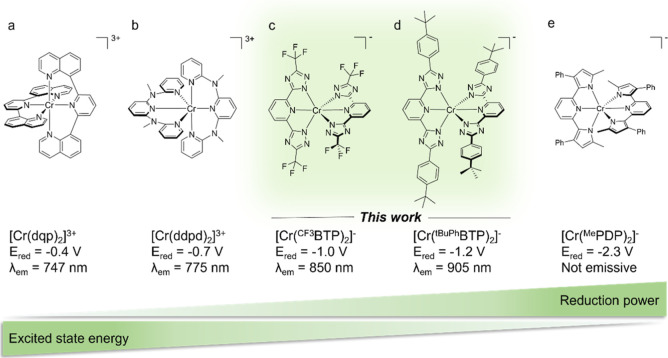
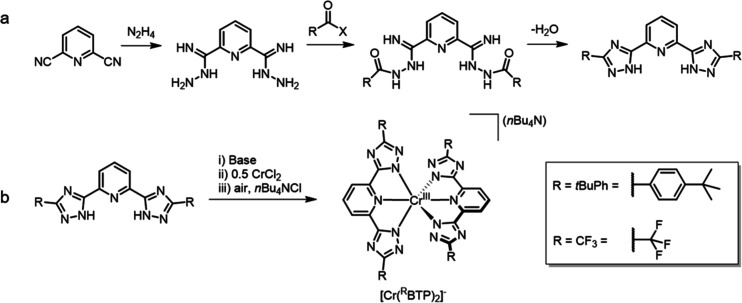
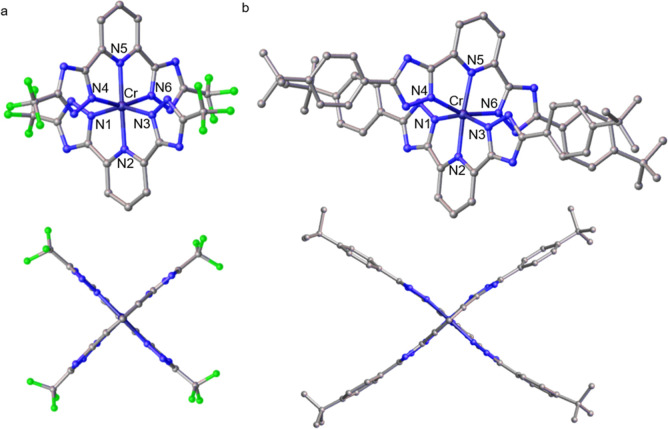
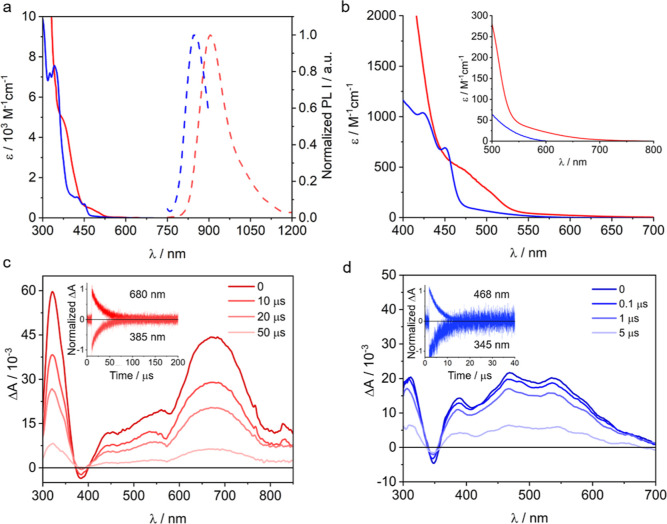
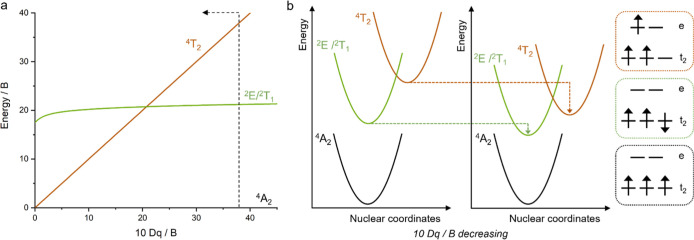
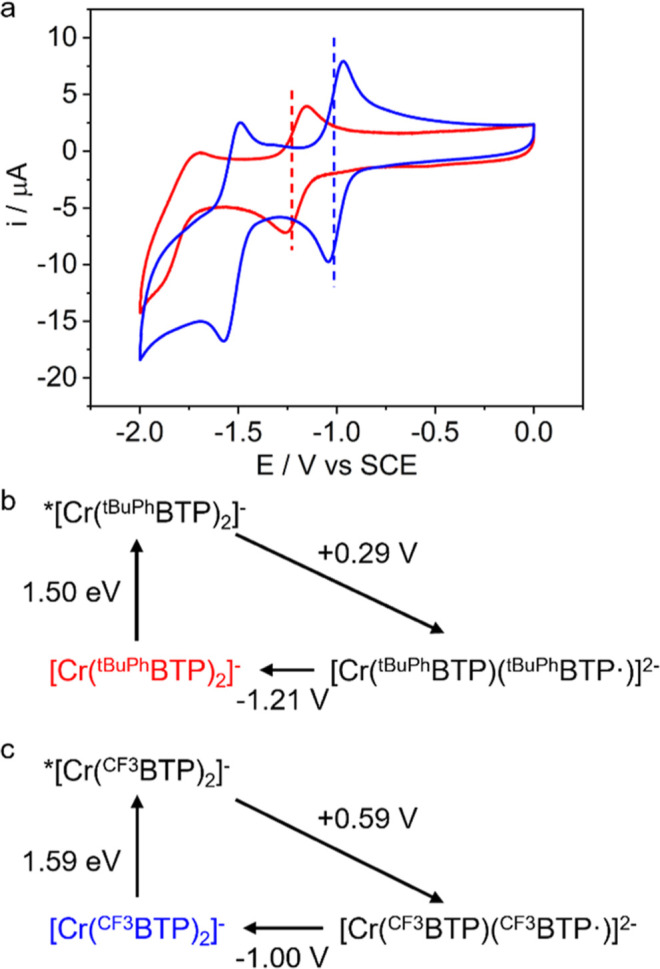
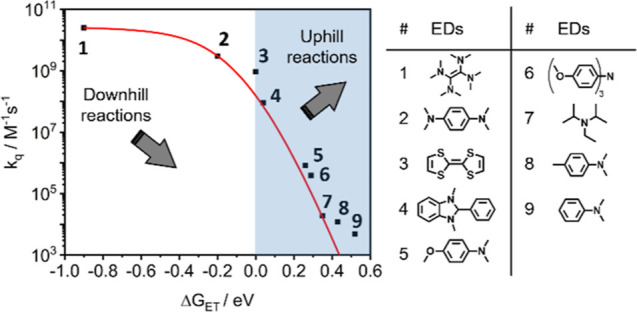
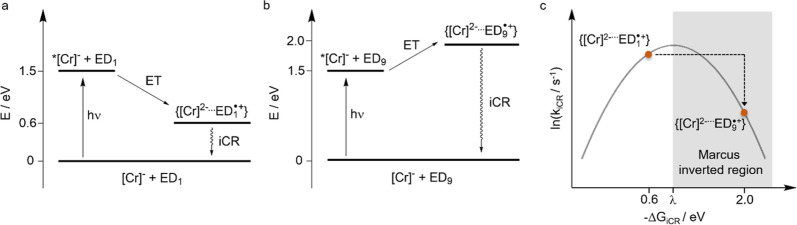
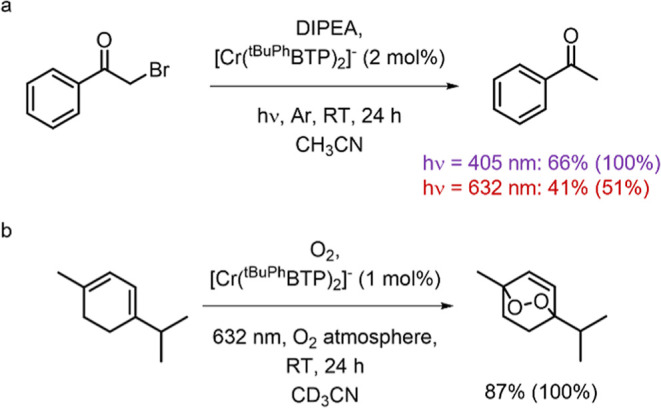
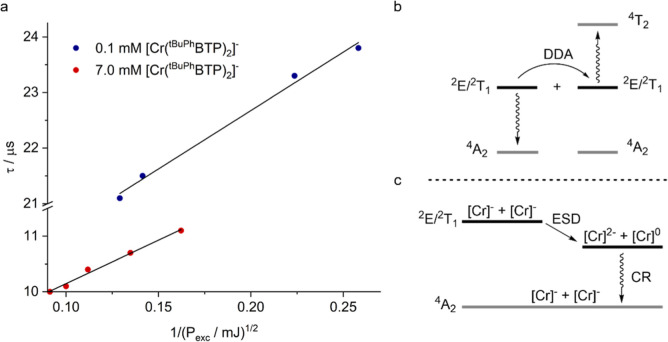
Photoactive CrIII complexes are typically based on polypyridine coordination environments, exhibit red luminescence, and are good photo-oxidants but have modest photoreducing properties. We report new CrIII complexes with anionic chelate ligands that enable color-tunable near-infrared luminescence and red-light-driven photoreduction reactions involving elementary steps that are endergonic up to 0.5 eV. Improving the metal-ligand bond covalency rather than more established approaches such as optimizing ligand field strength and coordination geometry is the underlying molecular design concept to achieve this favorable behavior. Our analysis suggests an intricate interplay between productive but slow endergonic photoinduced electron transfer and energy-wasting charge recombination rooted in cage escape effects, which could be generally important for photocatalysis. Our work also suggests the occurrence of doublet-doublet annihilation, a process that seems to have been largely neglected in current research on photoactive CrIII complexes but which could provide a mechanistic entry point into the widely used process of photochemical upconversion, typically based on triplet-triplet annihilation. Overall, this work conceptually advances the current state of the art of photoactive CrIII complexes in terms of molecular design, luminescence, and photoredox behavior. More generally, it informs photochemistry in terms of elucidating the limits of light-to-chemical energy conversion efficiency and the value of long-lived excited states in complexes of earth-abundant transition metals.
Figures
Similar articles
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
Sexual Harassment and Prevention Training.2024 Mar 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Mar 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 36508513 Free Books & Documents.
-
A rapid and systematic review of the clinical effectiveness and cost-effectiveness of paclitaxel, docetaxel, gemcitabine and vinorelbine in non-small-cell lung cancer.Health Technol Assess. 2001;5(32):1-195. doi: 10.3310/hta5320. Health Technol Assess. 2001. PMID: 12065068
-
The Black Book of Psychotropic Dosing and Monitoring.Psychopharmacol Bull. 2024 Jul 8;54(3):8-59. Psychopharmacol Bull. 2024. PMID: 38993656 Free PMC article. Review.
-
The Lived Experience of Autistic Adults in Employment: A Systematic Search and Synthesis.Autism Adulthood. 2024 Dec 2;6(4):495-509. doi: 10.1089/aut.2022.0114. eCollection 2024 Dec. Autism Adulthood. 2024. PMID: 40018061 Review.
References
-
- Kirk A. D., Porter G. B.. Luminescence of Chromium(III) Complexes. J. Phys. Chem. 1980;84:887–891. doi: 10.1021/j100445a020. - DOI
-
- Otto S., Dorn M., Förster C., Bauer M., Seitz M., Heinze K.. Understanding and exploiting long-lived near-infrared emission of a molecular ruby. Coord. Chem. Rev. 2018;359:102–111. doi: 10.1016/j.ccr.2018.01.004. - DOI
-
- Serpone N., Jamieson M. A., Henry M. S., Hoffman M. Z., Bolletta F., Maestri M.. Excited-State Behavior of Polypyridyl Complexes of Chromium(III) J. Am. Chem. Soc. 1979;101:2907–2916. doi: 10.1021/ja00505a019. - DOI
LinkOut - more resources
Full Text Sources
