

The unfolded protein response influences therapy outcome and disease progression in chronic lymphocytic leukaemia
- PMID: 40721481
- PMCID: PMC12304208
- DOI: 10.1038/s41598-025-13495-1
The unfolded protein response influences therapy outcome and disease progression in chronic lymphocytic leukaemia
Abstract
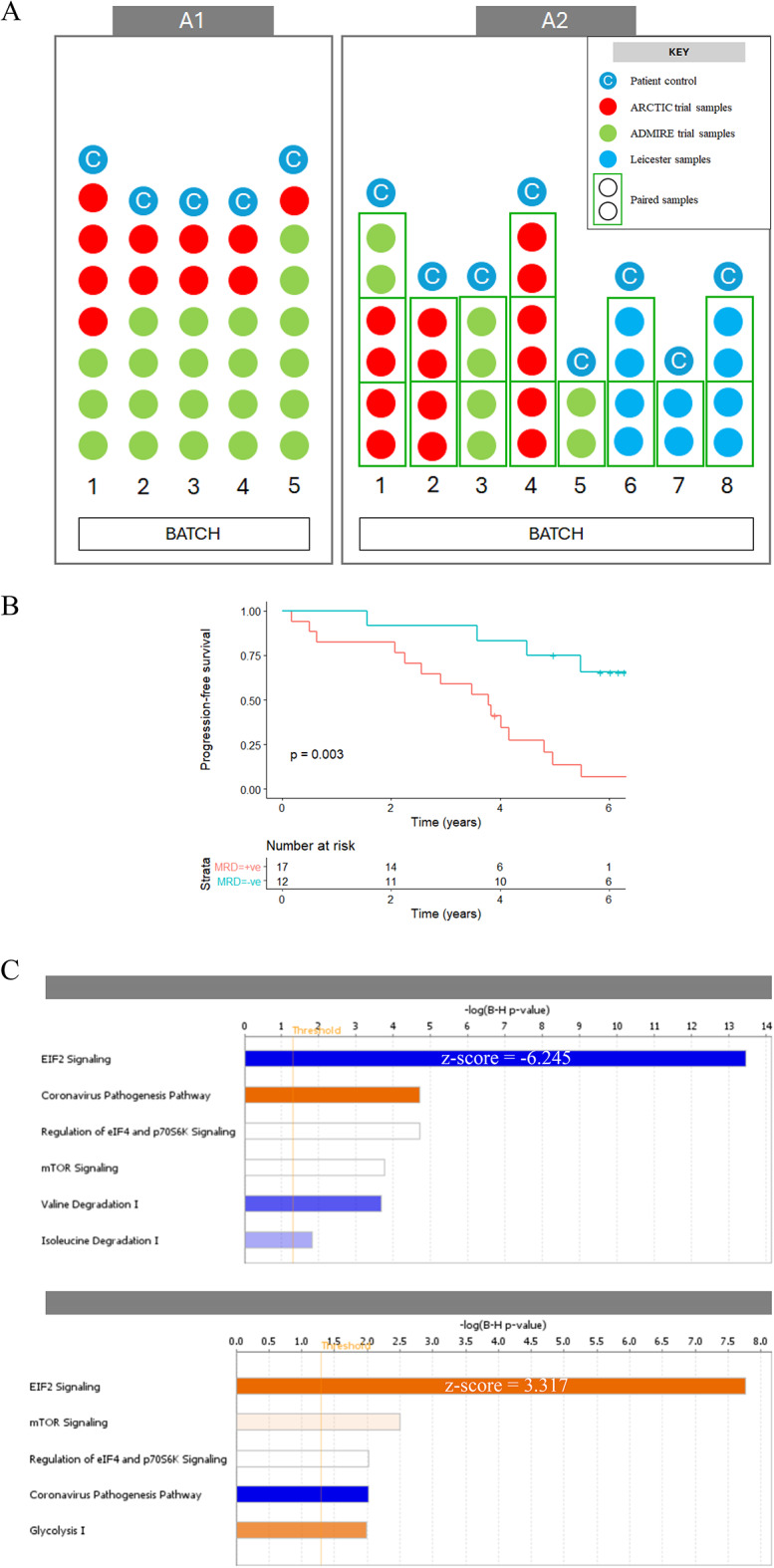
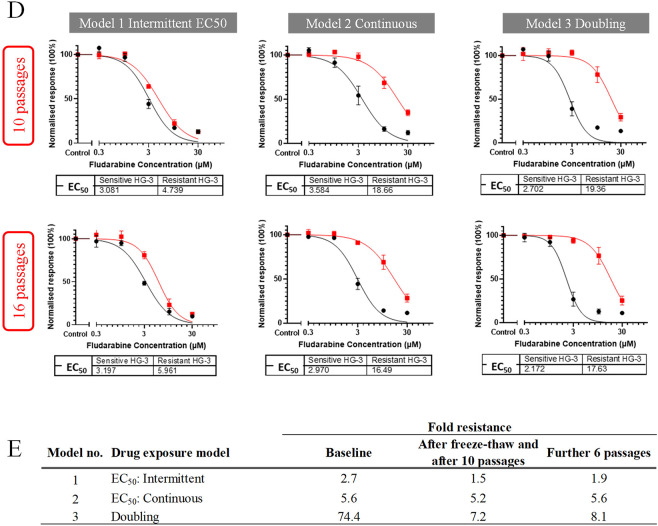
Since genomics, epigenomics and transcriptomics have provided only a partial explanation of chronic lymphocytic leukaemia (CLL) heterogeneity, and since concordance between mRNA and protein expression is incomplete, we related the CLL proteome to clinical outcome. CLL samples from patients who received fludarabine-containing chemoimmunotherapy were analysed by mass spectrometry (SWATH-MS). One dataset compared pre-treatment samples associated with an optimal versus suboptimal response, while another compared paired samples collected before treatment and at disease progression. eIF2 signalling (pivotal to the unfolded protein response (UPR)), was identified as the most enriched pathway in both datasets (respective z-scores: - 6.245 and 3.317; p < 0.0001), as well as in a fludarabine-resistant CLL cell line established from HG3 cells (z-score: - 2.121; p < 0.0001). Western blotting revealed that fludarabine-resistant HG3 cells expressed higher levels of PERK, which phosphorylates the regulatory eIF2α subunit, and lower levels of BiP, an HSP70 molecular chaperone that inactivates PERK but preferentially binds to misfolded proteins during ER stress. The PERK inhibitor, GSK2606414, sensitised resistant, but not sensitive, HG-3 cells to fludarabine without affecting background cell viability or cytotoxicity induced by the BCL-2 inhibitor venetoclax. These findings identify the UPR as a novel determinant of therapy outcome and disease progression in CLL.
Keywords: CLL cells; Drug resistance and disease progression; PERK linked to resistant phenotype; Proteomics; eIF2 signalling.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: UTK held a Fellowship that was partly funded by Eli Lilly, Novartis, Roche and UCB Pharma.AP has received research support/funding from Celgene/BMS, Gilead, GSK/Novartis, Napp, Roche. All the remaining authors declare no conflict of interest.
Figures





References
-
- Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute. Cancer Stat Facts: Leukemia—Chronic Lymphocytic Leukemia (CLL) – 2022, https://seer.cancer.gov/statfacts/html/clyl.html.
-
- Hallek, M. et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet376, 1164–1174. 10.1016/S0140-6736(10)61381-5 (2010). - PubMed
-
- Wierda, W. et al. Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab for relapsed and refractory chronic lymphocytic leukemia. J. Clin. Oncol.23, 4070–4078. 10.1200/JCO.2005.12.516 (2005). - PubMed
-
- Brown, J. R. The treatment of relapsed refractory chronic lymphocytic leukemia. Hematol. Am. Soc. Hematol. Educ. Program2011, 110–118 10.1182/asheducation-2011.1.110 (2011). - PubMed
-
- Zenz, T. et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood114, 2589–2597. 10.1182/blood-2009-05-224071 (2009). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
