

Deciphering state-dependent immune features from multi-layer omics data at single-cell resolution
- PMID: 40721531
- PMCID: PMC12339405
- DOI: 10.1038/s41588-025-02266-3
Deciphering state-dependent immune features from multi-layer omics data at single-cell resolution
Abstract
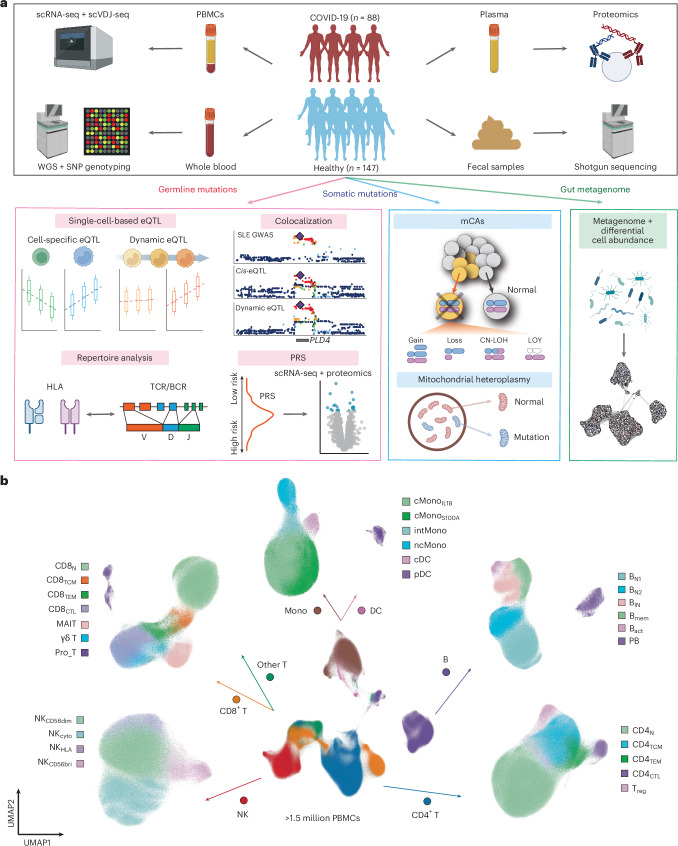
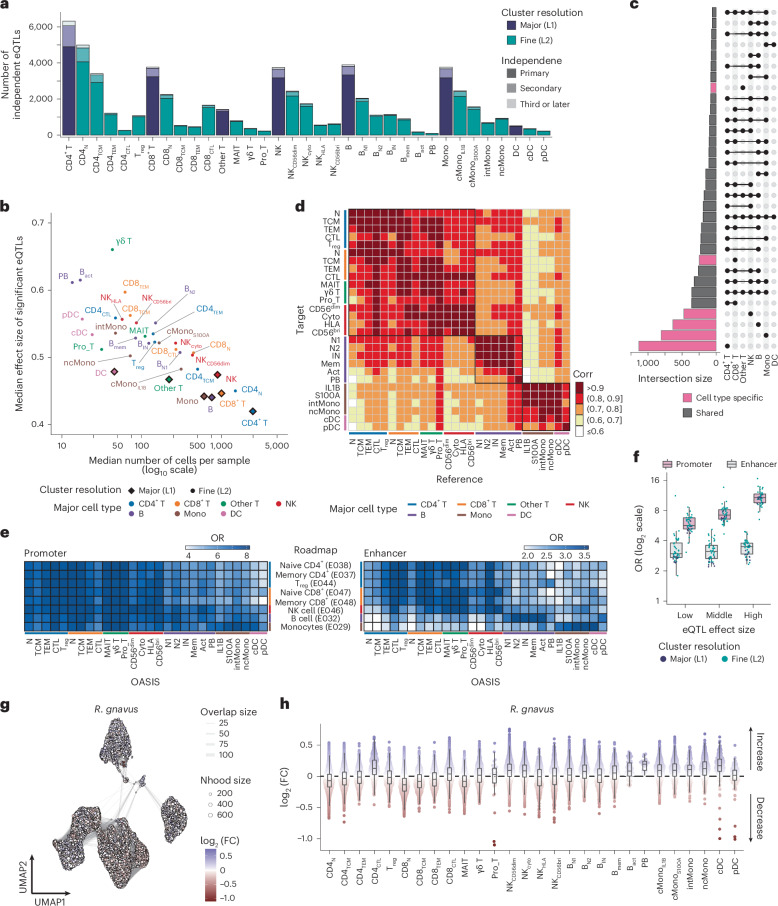
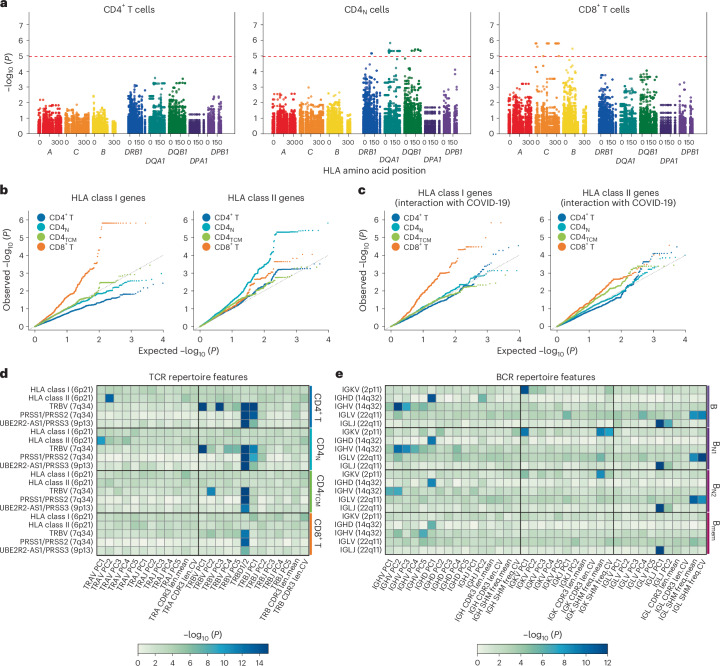
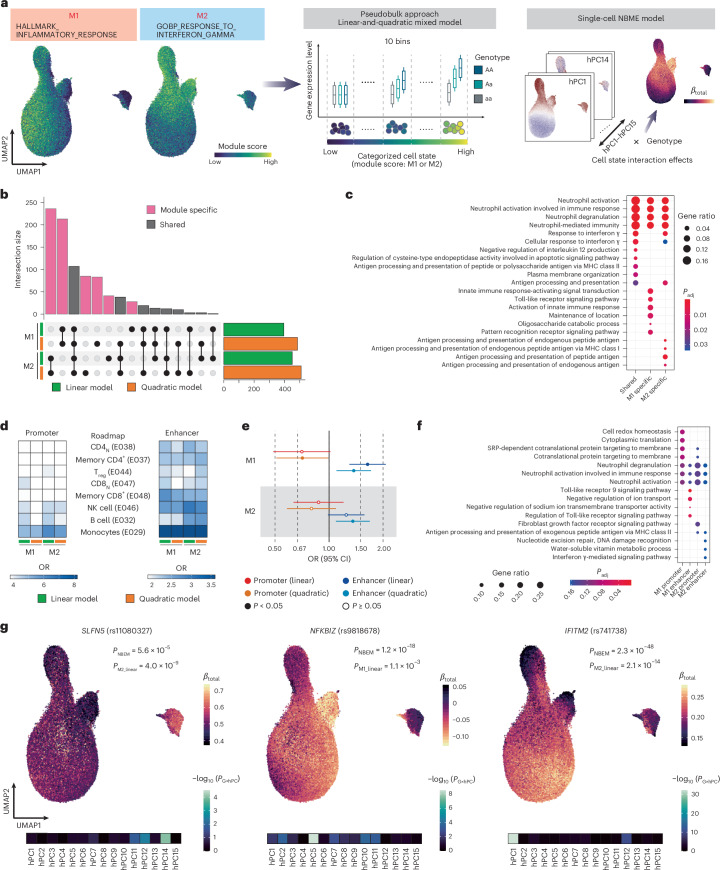
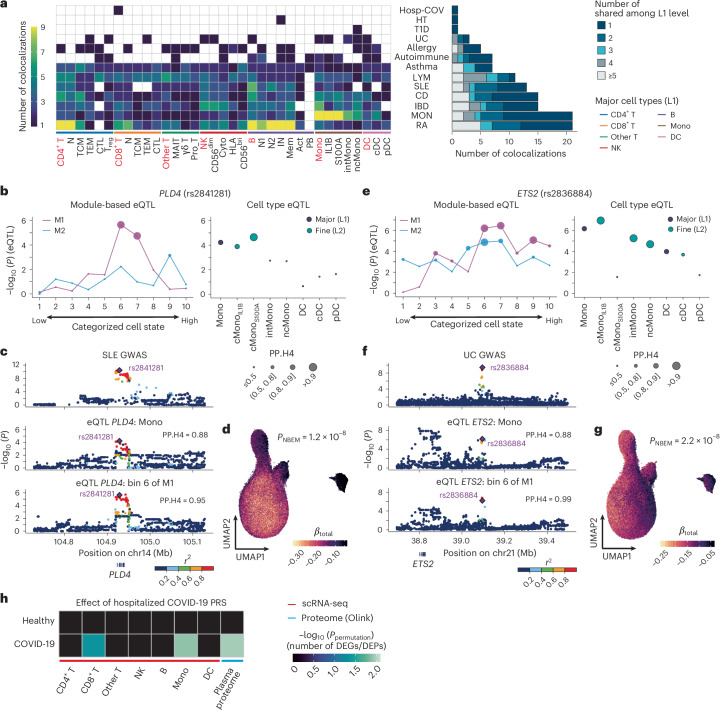
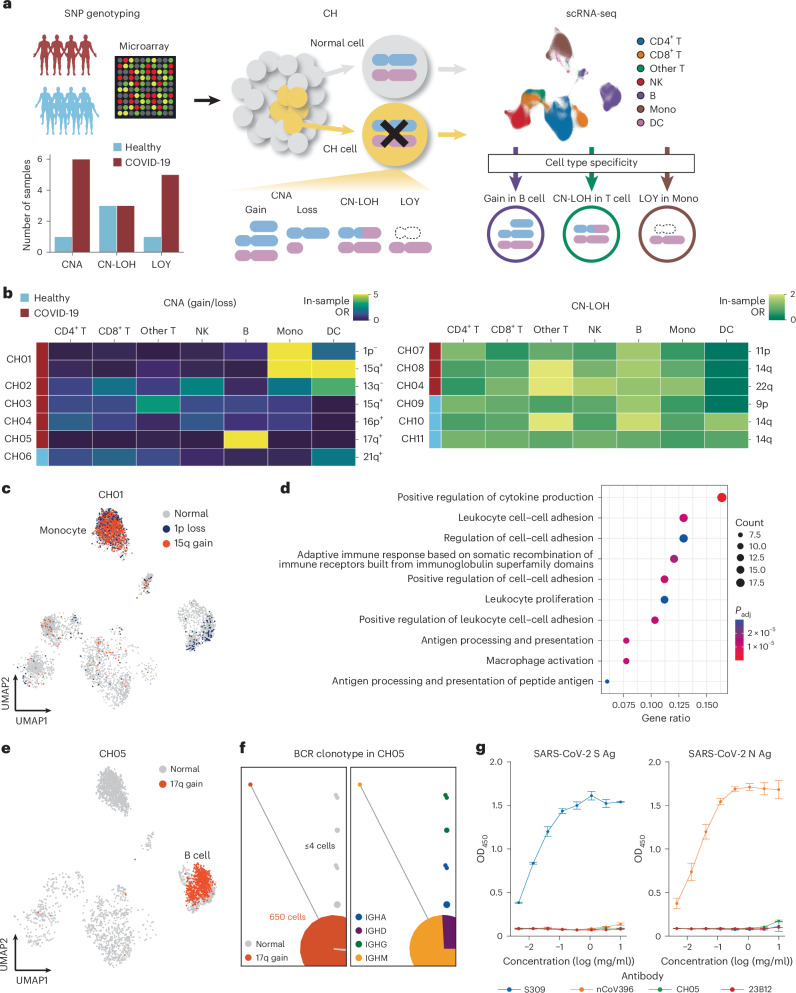
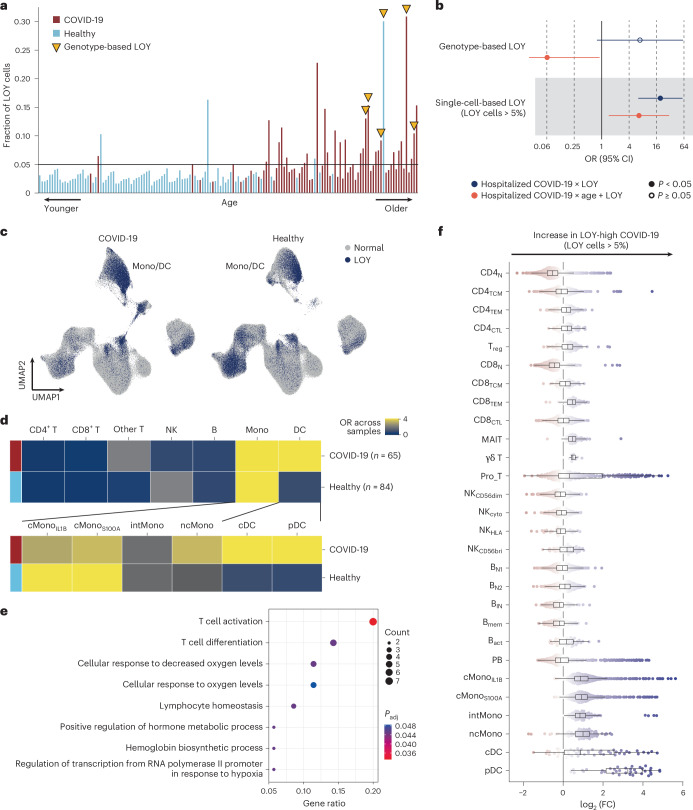
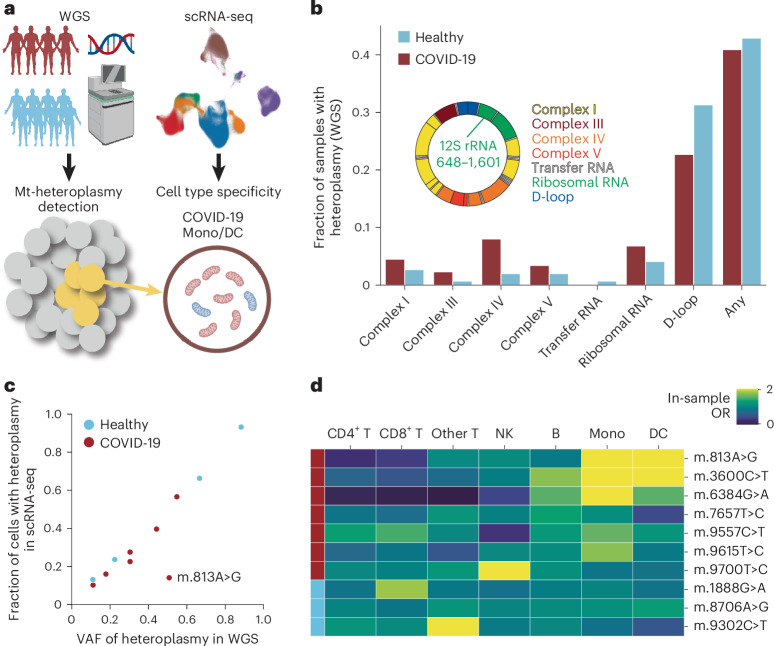
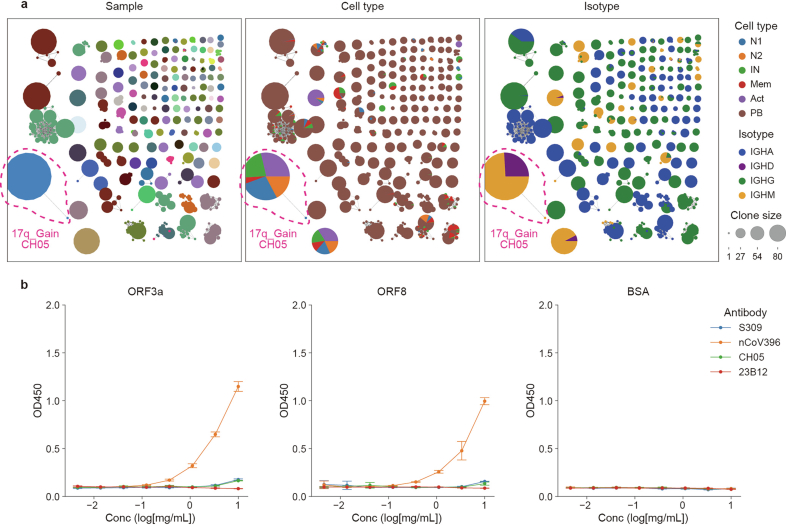
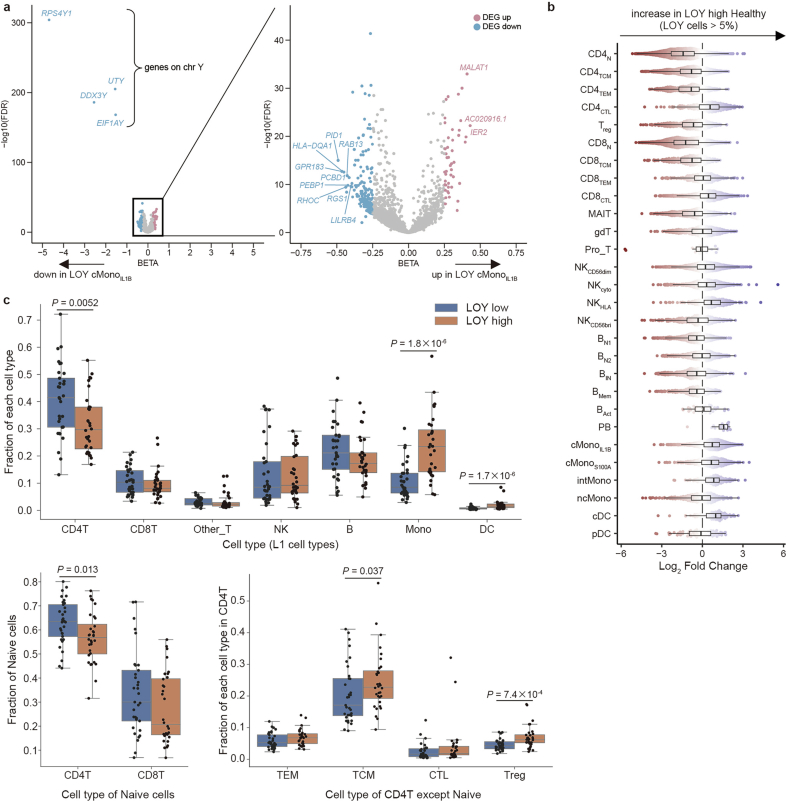
Current molecular quantitative trait locus catalogs are mostly at bulk resolution and centered on Europeans. Here, we constructed an immune cell atlas with single-cell transcriptomics of >1.5 million peripheral blood mononuclear cells, host genetics, plasma proteomics and gut metagenomics from 235 Japanese persons, including patients with coronavirus disease 2019 (COVID-19) and healthy individuals. We mapped germline genetic effects on gene expression within immune cell types and across cell states. We elucidated cell type- and context-specific human leukocyte antigen (HLA) and genome-wide associations with T and B cell receptor repertoires. Colocalization using dynamic genetic regulation provided better understanding of genome-wide association signals. Differential gene and protein expression analyses depicted cell type- and context-specific effects of polygenic risks. Various somatic mutations including mosaic chromosomal alterations, loss of Y chromosome and mitochondrial DNA (mtDNA) heteroplasmy were projected into single-cell resolution. We identified immune features specific to somatically mutated cells. Overall, immune cells are dynamically regulated in a cell state-dependent manner characterized with multiomic profiles.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
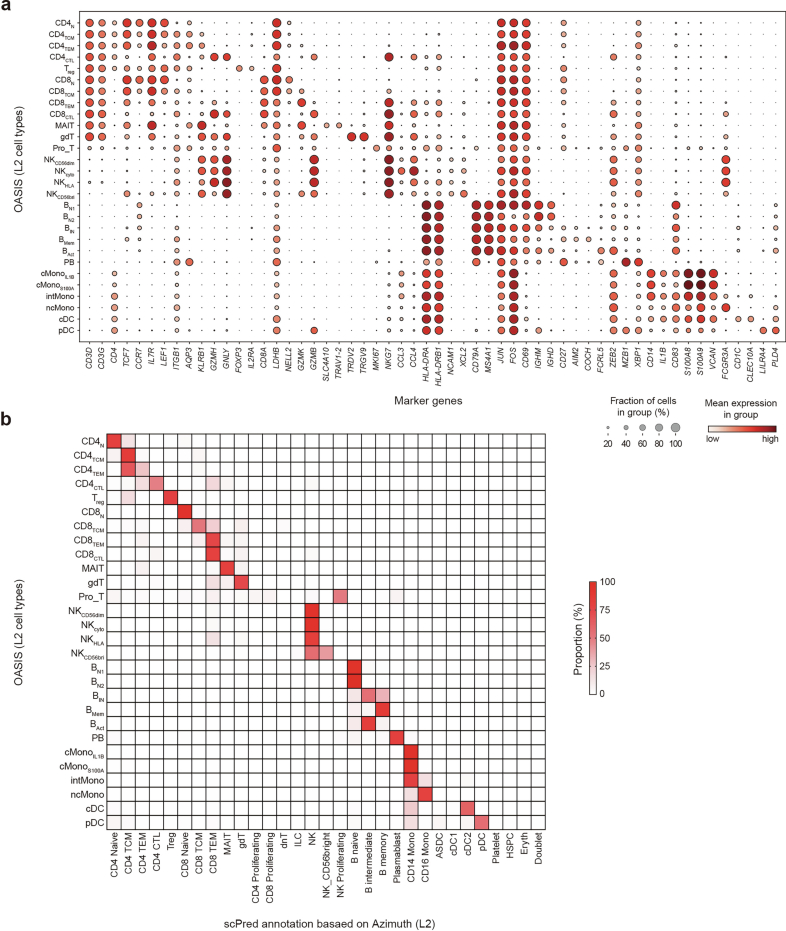
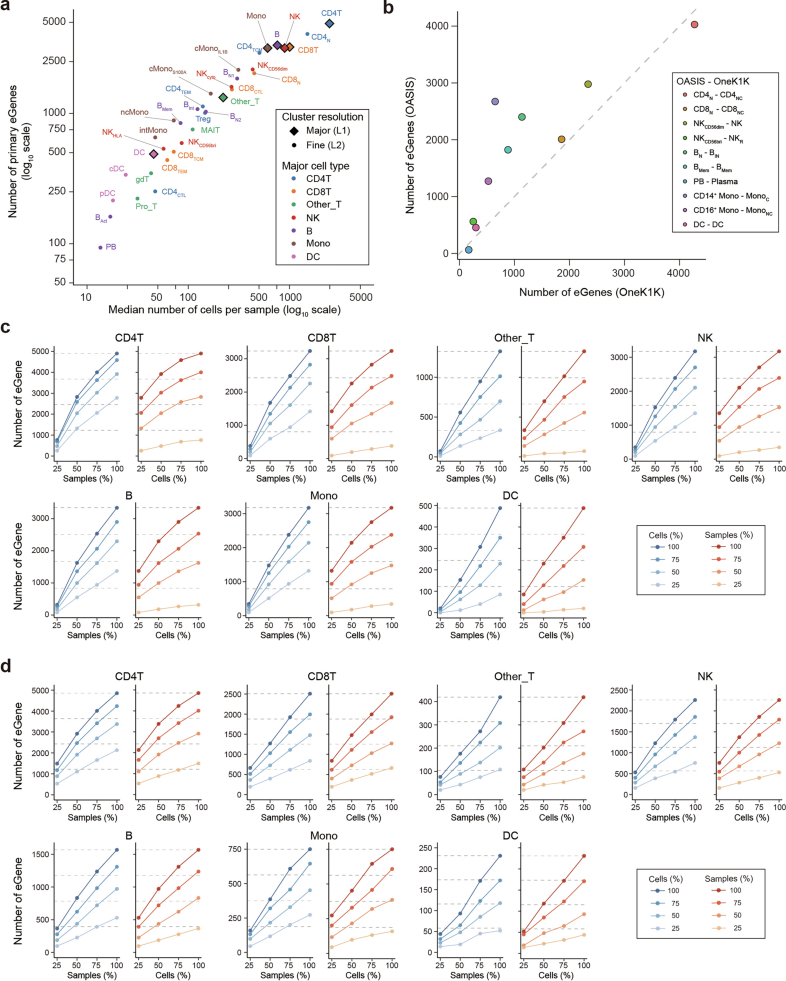
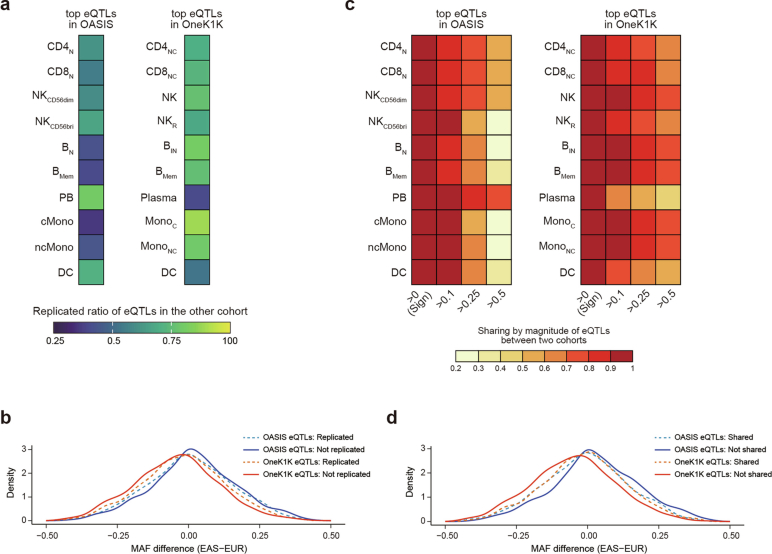
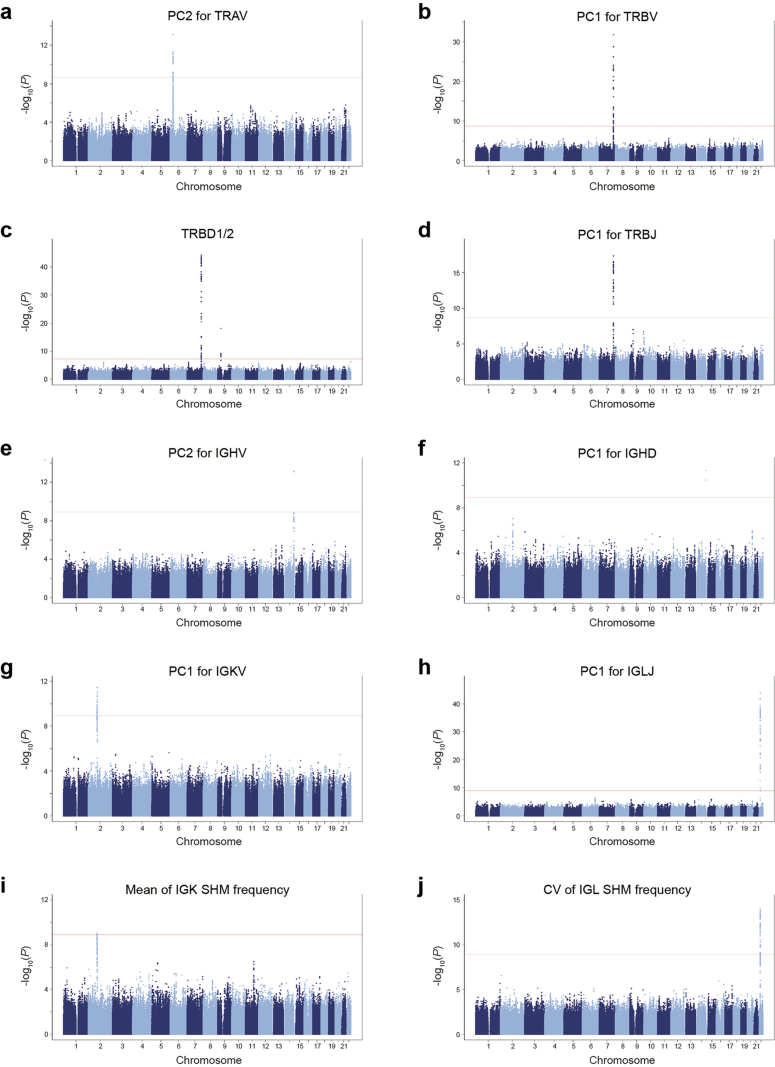
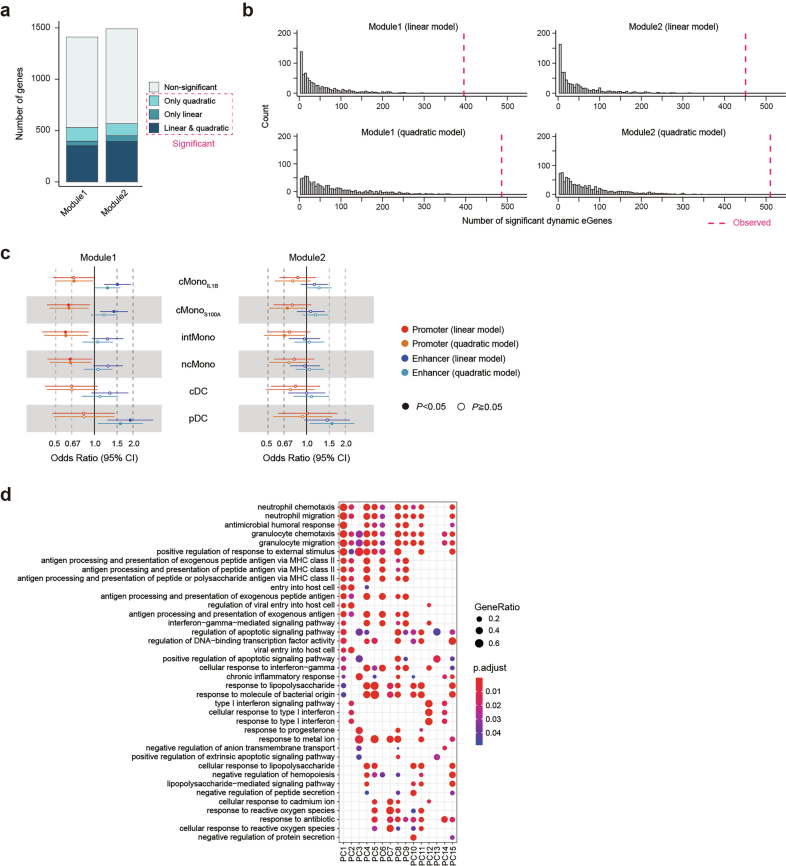
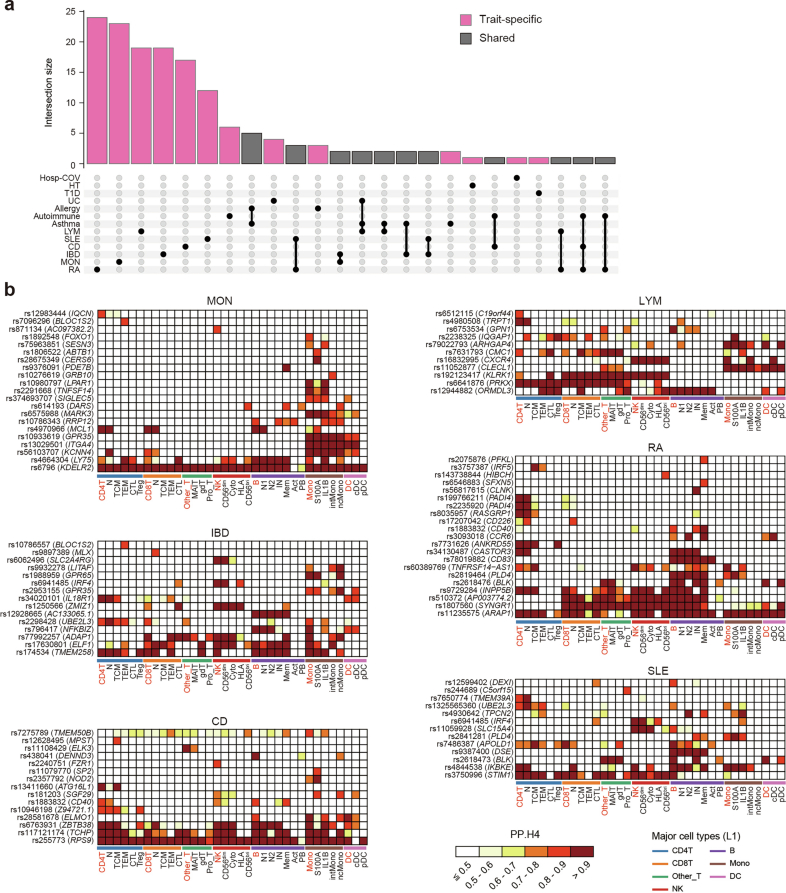
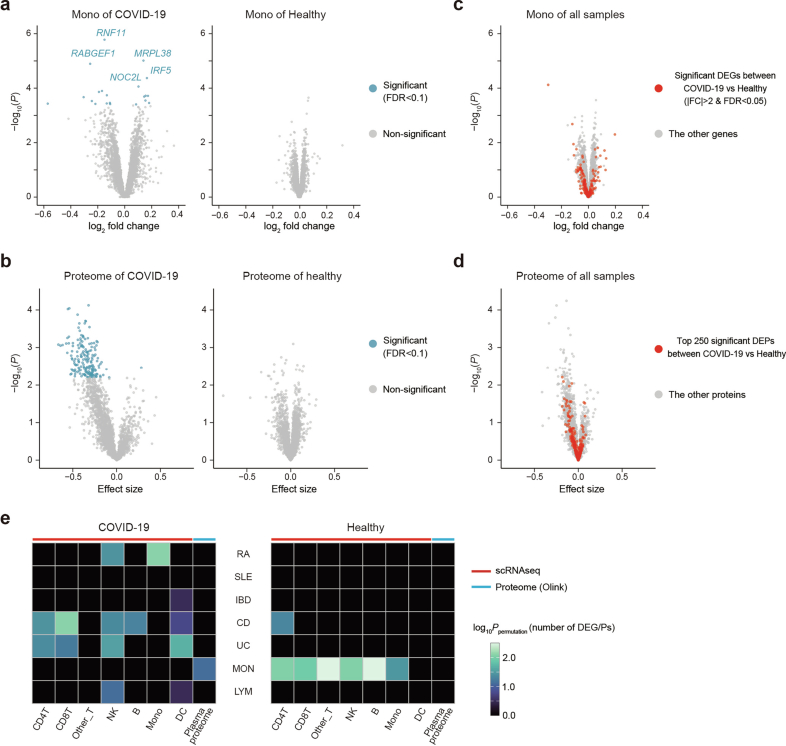
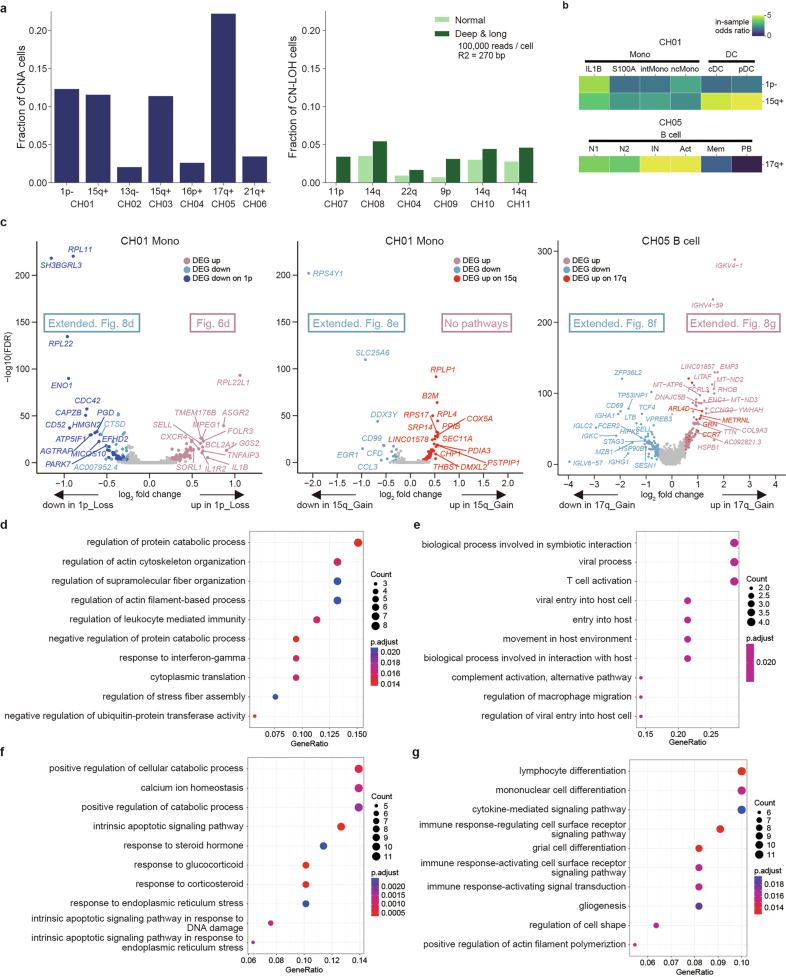
References
-
- Aguet, F. et al. Molecular quantitative trait loci. Nat. Rev. Methods Primers3, 5 (2023).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
