

Modulation of Redox-Sensitive Cardiac Ion Channels
- PMID: 40722941
- PMCID: PMC12291642
- DOI: 10.3390/antiox14070836
Modulation of Redox-Sensitive Cardiac Ion Channels
Abstract
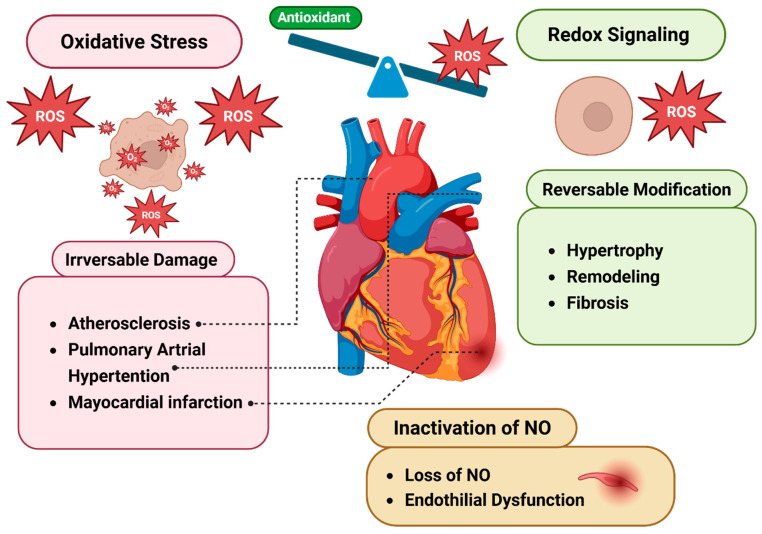
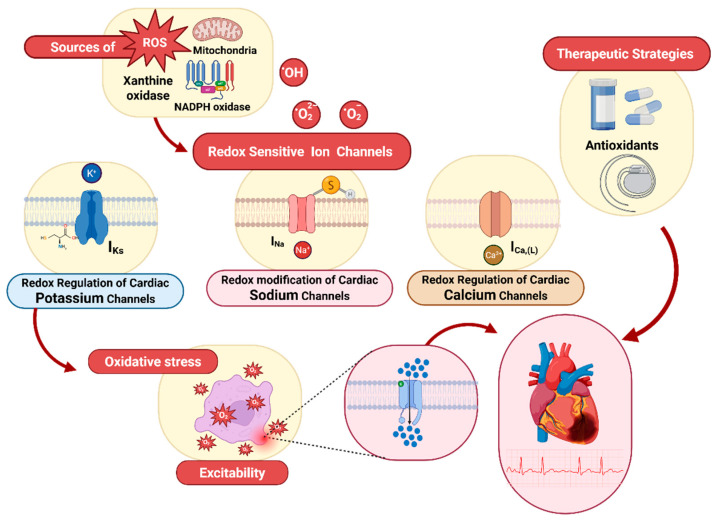
Redox regulation is crucial for the cardiac action potential, coordinating the sodium-driven depolarization, calcium-mediated plateau formation, and potassium-dependent repolarization processes required for proper heart function. Under physiological conditions, low-level reactive oxygen species (ROS), generated by mitochondria and membrane oxidases, adjust ion channel function and support excitation-contraction coupling. However, when ROS accumulate, they modify a variety of important channel proteins in cardiomyocytes, which commonly results in reducing potassium currents, enhancing sodium and calcium influx, and enhancing intracellular calcium release. These redox-driven alterations disrupt the cardiac rhythm, promote after-depolarizations, impair contractile force, and accelerate the development of heart diseases. Experimental models demonstrate that oxidizing agents reduce repolarizing currents, whereas reducing systems restore normal channel activity. Similarly, oxidative modifications of calcium-handling proteins amplify sarcoplasmic reticulum release and diastolic calcium leak. Understanding the precise redox-dependent modifications of cardiac ion channels would guide new possibilities for targeted therapies aimed at restoring electrophysiological homeostasis under oxidative stress, potentially alleviating myocardial infarction and cardiovascular dysfunction.
Keywords: antioxidants; cardiomyocytes; cardiovascular diseases; heart failure; ion channels; kinase; mitochondria; oxidative stress; reactive oxygen species; redox signaling.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures




References
-
- Mandal M., Sarkar M., Khan A., Biswas M., Masi A., Rakwal R., Agrawal G.K., Srivastava A., Sarkar A. Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS) in plants—Maintenance of structural individuality and functional blend. Adv. Redox Res. 2022;5:100039. doi: 10.1016/j.arres.2022.100039. - DOI
-
- Juan C.A., Pérez de la Lastra J.M., Plou F.J., Pérez-Lebeña E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021;22:4642. doi: 10.3390/ijms22094642. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
