

Retinoic Acid Induced 1 and Smith-Magenis Syndrome: From Genetics to Biology and Possible Therapeutic Strategies
- PMID: 40724914
- PMCID: PMC12296005
- DOI: 10.3390/ijms26146667
Retinoic Acid Induced 1 and Smith-Magenis Syndrome: From Genetics to Biology and Possible Therapeutic Strategies
Abstract
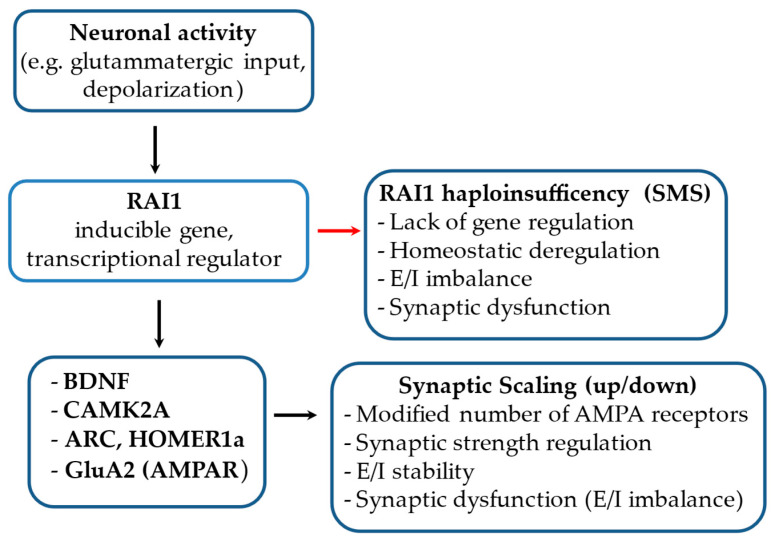
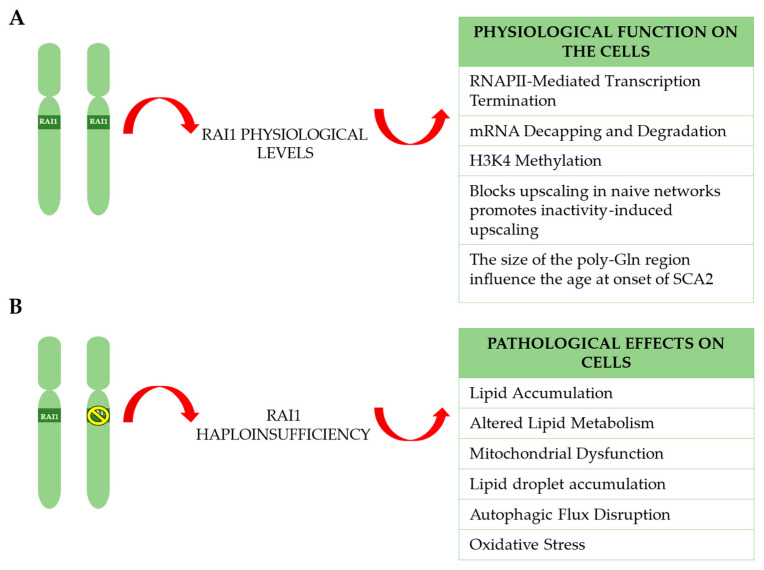
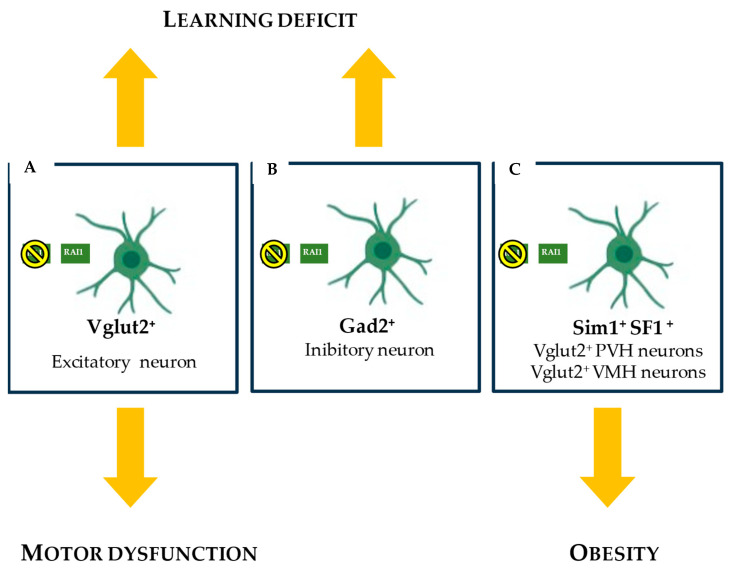
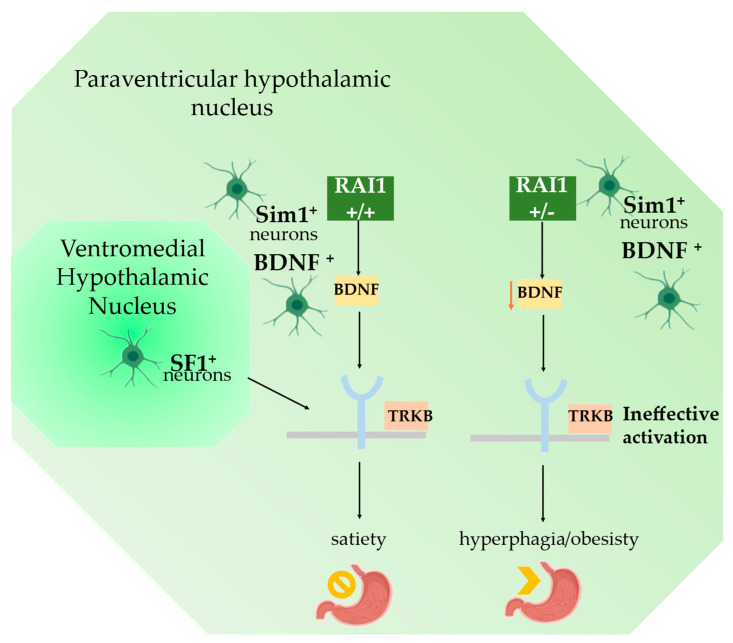
Haploinsufficiency disorders are genetic diseases caused by reduced gene expression, leading to developmental, metabolic, and tumorigenic abnormalities. The dosage-sensitive Retinoic Acid Induced 1 (RAI1) gene, located within the 17p11.2 region, is central to the core features of Smith--Magenis syndrome (SMS) and Potocki--Lupski syndrome (PTLS), caused by the reciprocal microdeletions and microduplications of this region, respectively. SMS and PTLS present contrasting phenotypes. SMS is characterized by severe neurobehavioral manifestations, sleep disturbances, and metabolic abnormalities, and PTLS shows milder features. Here, we detail the molecular functions of RAI1 in its wild-type and haploinsufficiency conditions (RAI1+/-), as studied in animal and cellular models. RAI1 acts as a transcription factor critical for neurodevelopment and synaptic plasticity, a chromatin remodeler within the Histone 3 Lysine 4 (H3K4) writer complex, and a regulator of faulty 5'-capped pre-mRNA degradation. Alterations of RAI1 functions lead to synaptic scaling and transcriptional dysregulation in neural networks. This review highlights key molecular mechanisms of RAI1, elucidating its role in the interplay between genetics and phenotypic features and summarizes innovative therapeutic approaches for SMS. These data provide a foundation for potential therapeutic strategies targeting RAI1, its mRNA products, or downstream pathways.
Keywords: HD; PTLS; RAI1; SMS.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

Similar articles
-
Retinoic Acid-Induced 1 Gene and Neuropsychiatric Diseases: A Systematic Review.Expert Rev Mol Med. 2025 May 29;27:e17. doi: 10.1017/erm.2025.12. Expert Rev Mol Med. 2025. PMID: 40437981 Free PMC article. Review.
-
Generation and characterization of the CSSi021-A (15665) human induced pluripotent stem cell line from a Smith-Magenis syndrome patient with a heterozygous RAI1 mutation.Stem Cell Res. 2025 Aug;86:103726. doi: 10.1016/j.scr.2025.103726. Epub 2025 Apr 26. Stem Cell Res. 2025. PMID: 40311325
-
Smith-Magenis syndrome: haploinsufficiency of RAI1 results in altered gene regulation in neurological and metabolic pathways.Expert Rev Mol Med. 2011 Apr 19;13:e14. doi: 10.1017/S1462399411001827. Expert Rev Mol Med. 2011. PMID: 21545756 Review.
-
Potocki-Lupski Syndrome.2017 Aug 24. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2017 Aug 24. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 28837307 Free Books & Documents. Review.
-
Molecular analysis of the Retinoic Acid Induced 1 gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion.PLoS One. 2011;6(8):e22861. doi: 10.1371/journal.pone.0022861. Epub 2011 Aug 8. PLoS One. 2011. PMID: 21857958 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
