

Discordant effects of maternal age on the human MII oocyte transcriptome
- PMID: 40729310
- PMCID: PMC12360849
- DOI: 10.1093/molehr/gaaf038
Discordant effects of maternal age on the human MII oocyte transcriptome
Abstract
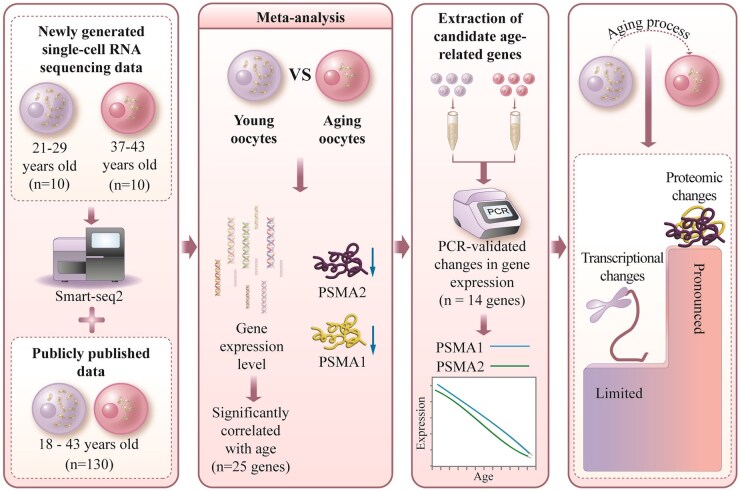
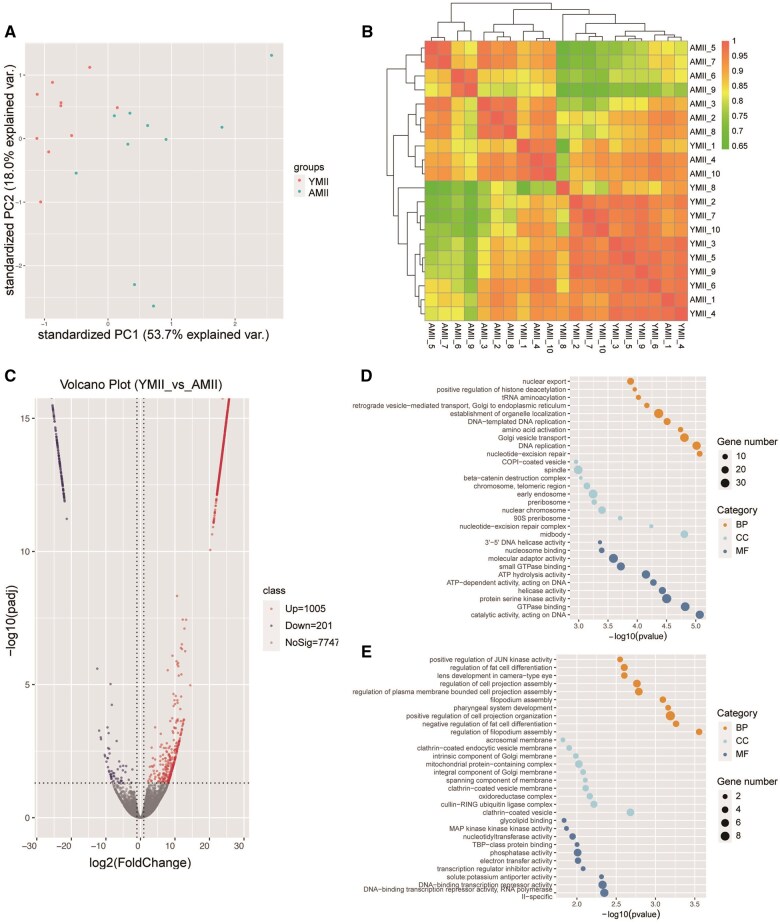
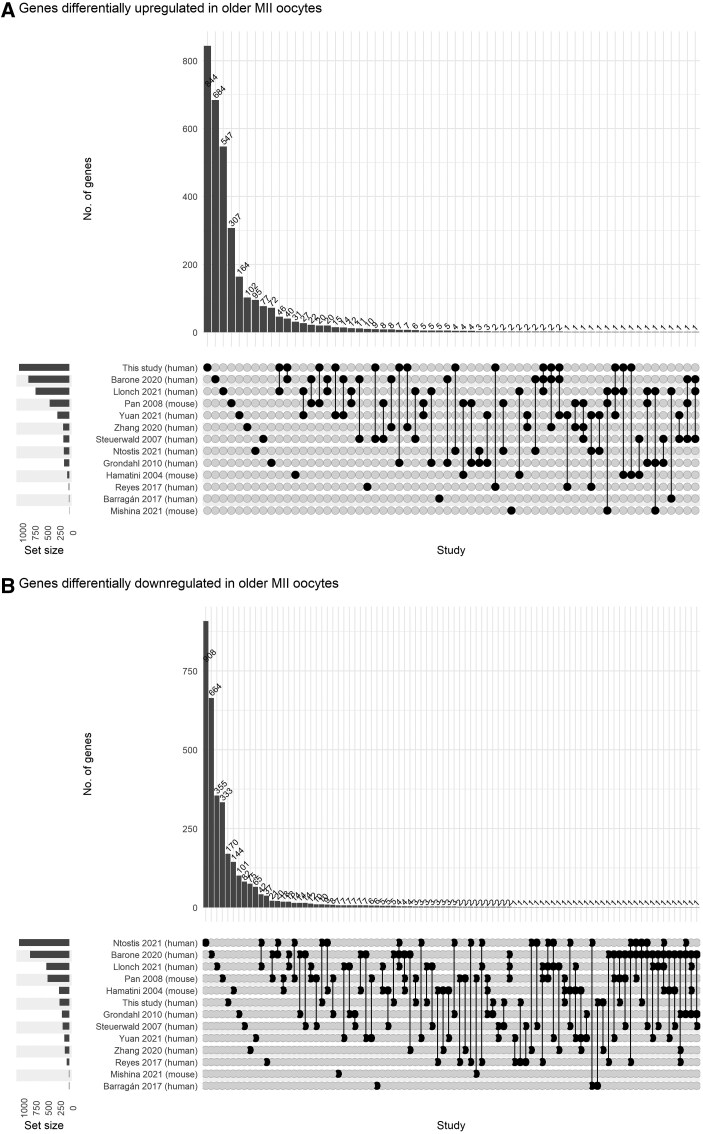
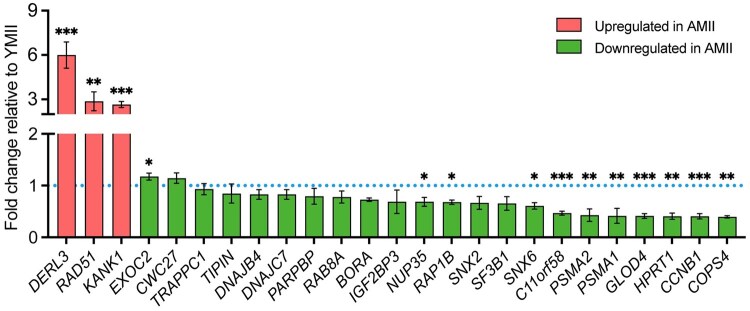
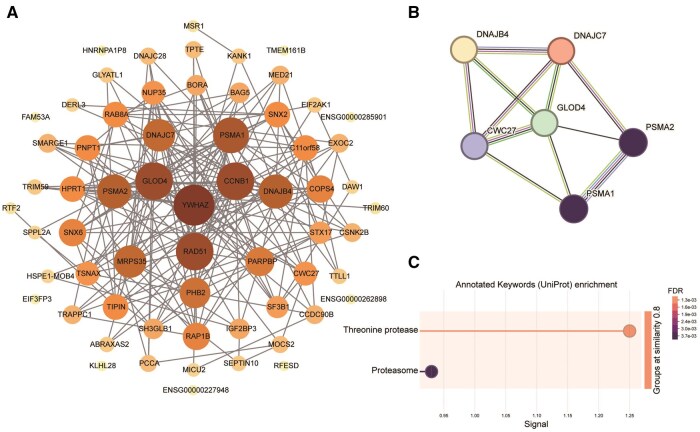
While advanced maternal age is associated with significant changes in oocyte gene expression, these are not global changes but limited to a fraction of the transcriptome. However, there is little consensus on the specific genes affected, and on the transcriptomic signatures of age-related declines in oocyte quality. To characterize the effects of age on the human MII oocyte transcriptome, here we take a two-part approach. We first generated single-oocyte Smart-seq2 datasets from 10 younger (21-29 years) and 10 older (37-43 years) donors, identifying genes differentially expressed between the two groups, then cross-referenced our results with those of 12 studies (9 human, 3 mouse) performing equivalent analyses using a variety of single-cell transcriptomic or microarray platforms. Technical differences notwithstanding, we found considerable discordance between the datasets, suggesting that age-related signatures of differential gene expression are not easily reproducible. Independent corroboration of age-associated changes in expression was limited to few genes, with the vast majority only supported by one of the 13 datasets, including our own. Nevertheless, we identified 40 genes whose expression significantly altered with age in multiple studies, highlighting common processes underlying ageing, including dysregulated proteostasis. As human Smart-seq2 oocyte libraries are challenging to procure and rare in public archives, we next implemented a meta-analytic method for their re-use, combining our 20 oocytes with 130 pre-existing libraries sourced from 12 different studies and representing a continuous age range of 18-43 years. We identified 25 genes whose expression level significantly correlated with age and corroborated 14 of these genes with RT-PCR, including the proteasomal subunits PSMA1 and PSMA2, both of which were downregulated in older oocytes. Overall, our findings are consistent with both pronounced inter-oocyte heterogeneity in transcription and with oocyte ageing being a multifactorial process to which bona fide transcriptomic changes may only play a restricted role, while proteomic changes play more pronounced roles.
Keywords: differential gene expression; meta-analysis; oocyte ageing; oocyte quality; proteasome.
© The Author(s) 2025. Published by Oxford University Press on behalf of European Society of Human Reproduction and Embryology.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
Similar articles
-
Impact of aging on gene expression in human oocytes: a comparative analysis of young and older patients.Reprod Biol Endocrinol. 2025 Jul 29;23(1):111. doi: 10.1186/s12958-025-01449-1. Reprod Biol Endocrinol. 2025. PMID: 40731018 Free PMC article.
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Single-cell analysis comparing early-stage oocytes from fresh and slow-frozen/thawed human ovarian cortex reveals minimal impact of cryopreservation on the oocyte transcriptome.Hum Reprod. 2025 Apr 1;40(4):683-694. doi: 10.1093/humrep/deaf009. Hum Reprod. 2025. PMID: 39919251
-
Systemic treatments for metastatic cutaneous melanoma.Cochrane Database Syst Rev. 2018 Feb 6;2(2):CD011123. doi: 10.1002/14651858.CD011123.pub2. Cochrane Database Syst Rev. 2018. PMID: 29405038 Free PMC article.
-
Falls prevention interventions for community-dwelling older adults: systematic review and meta-analysis of benefits, harms, and patient values and preferences.Syst Rev. 2024 Nov 26;13(1):289. doi: 10.1186/s13643-024-02681-3. Syst Rev. 2024. PMID: 39593159 Free PMC article.
References
-
- Asghari A, Marashi S-A, Ansari-Pour N. A sperm-specific proteome-scale metabolic network model identifies non-glycolytic genes for energy deficiency in asthenozoospermia. Syst Biol Reprod Med 2017;63:100–112. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
