

Enabling scalable single-cell transcriptomic analysis through distributed computing with Apache spark
- PMID: 40731055
- PMCID: PMC12307815
- DOI: 10.1038/s41598-025-12897-5
Enabling scalable single-cell transcriptomic analysis through distributed computing with Apache spark
Abstract
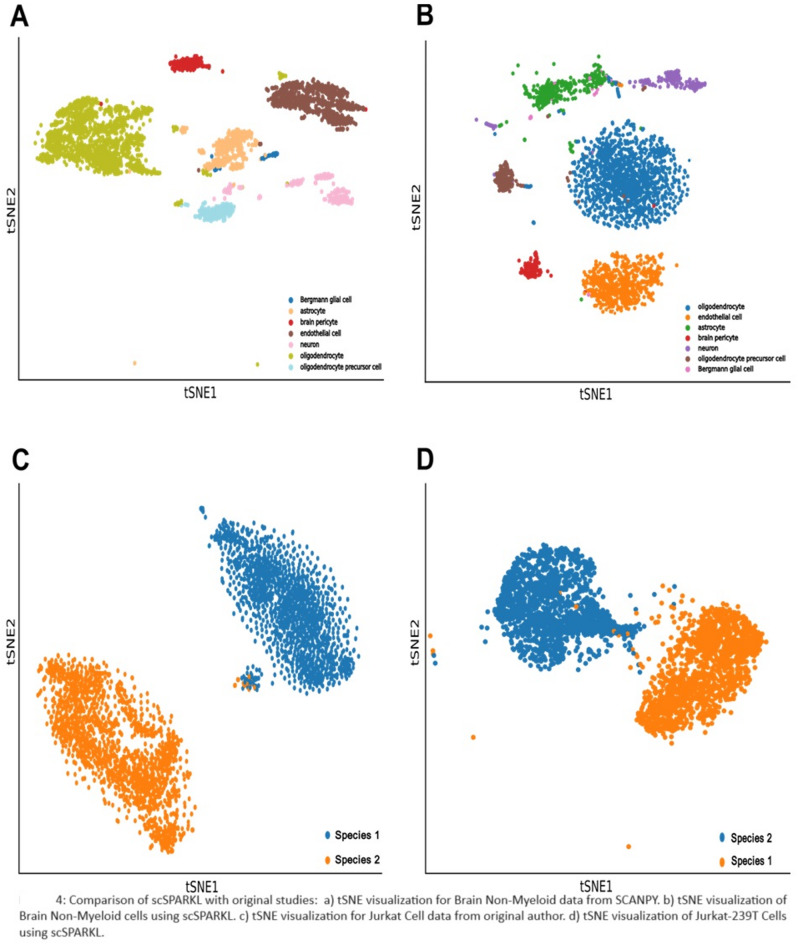
As the field of single-cell genomics continues to develop, the generation of large-scale scRNA-seq datasets has become more prevalent. Although these datasets offer tremendous potential for shedding light on the complex biology of individual cells, the sheer volume of data presents significant challenges for management and analysis. Off late, to address these challenges, a new discipline, known as "big single-cell data science," has emerged. Within this field, a variety of computational tools have been developed to facilitate the processing and interpretation of scRNA-seq data. However, several of these tools primarily focus on the analytical aspect and tend to overlook the burgeoning data deluge generated by scRNA-seq experiments. In this study, we try to address this challenge and present a novel parallel analytical framework, scSPARKL, that leverages the power of Apache Spark to enable the efficient analysis of single-cell transcriptomic data. scSPARKL is fortified by a rich set of staged algorithms developed to optimize the Apache Spark's work environment. The tool incorporates six key operations for dealing with single-cell Big Data, including data reshaping, data preprocessing, cell/gene filtering, data normalization, dimensionality reduction, and clustering. By utilizing Spark's unlimited scalability, fault tolerance, and parallelism, the tool enables researchers to rapidly and accurately analyze scRNA-seq datasets of any size. We demonstrate the utility of our framework and algorithms through a series of experiments on real-world scRNA-seq data. Overall, our results suggest that scSPARKL represents a powerful and flexible tool for the analysis of single-cell transcriptomic data, with broad applications across the fields of biology and medicine.
Keywords: Apache spark; Big data; Normalization; Quality control; ScRNA-seq.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests.
Figures




References
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
