

Bridging the Gap in Breast Cancer Dormancy: Models, Mechanisms, and Translational Challenges
- PMID: 40732251
- PMCID: PMC12298558
- DOI: 10.3390/ph18070961
Bridging the Gap in Breast Cancer Dormancy: Models, Mechanisms, and Translational Challenges
Abstract
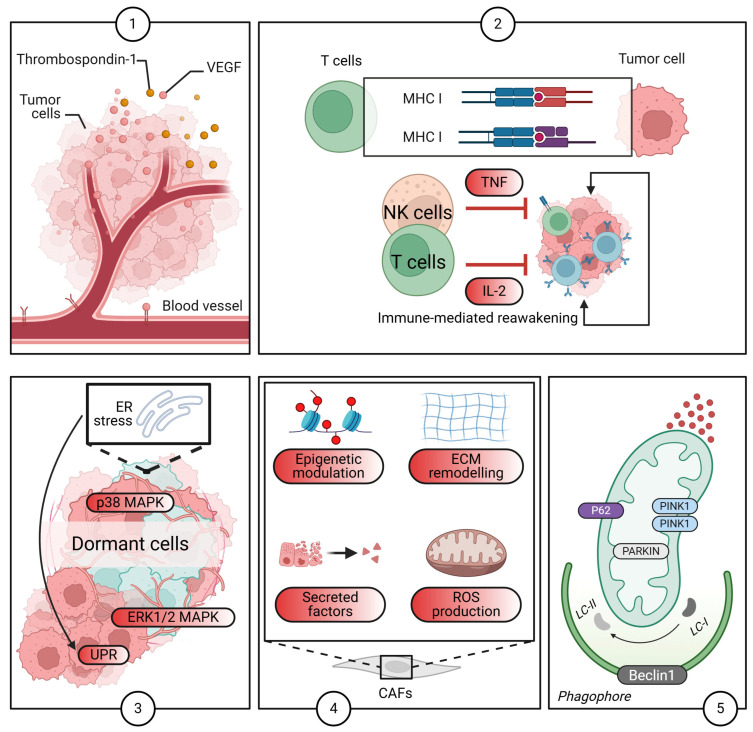
Breast cancer (BC) poses a significant clinical challenge due to late metastatic recurrence, driven by dormant disseminated tumor cells (DTCs). This review emphasizes the urgency of addressing tumor dormancy to reduce metastatic relapse, a major contributor to BC mortality. DTCs evade conventional therapies and immune surveillance, reactivating unpredictably, thus necessitating targeted strategies. Current research is fragmented, with conflicting data, inadequate models, and a lack of biomarkers hindering progress. This review synthesizes these gaps and proposes actionable priorities, advocating for integrated, standardized approaches. It highlights the roles of single-cell multi-omics, spatial transcriptomics, and humanized long-term models in unraveling dormancy mechanisms. The review also emphasizes macrophage-targeted therapies, dormancy-specific trials, and biomarker validation, offering paths to clinical translation. Ultimately, this work emphasizes the urgent need for integrated multi-omics approaches, including single-cell and spatial transcriptomics, combined with advanced computational analysis. Moreover, this review critically analyzes the existing research landscape, meticulously identifying key gaps, and proposing concrete, forward-looking directions for both fundamental research and clinical translation in the challenging field of BC dormancy.
Keywords: BC; metastatic recurrence; microenvironment; translational research; tumor dormancy.
Conflict of interest statement
All authors declare no conflicts of interests.
Figures




Similar articles
-
Decoding the adaptive survival mechanisms of breast cancer dormancy.Oncogene. 2025 Aug 27. doi: 10.1038/s41388-025-03529-3. Online ahead of print. Oncogene. 2025. PMID: 40866498 Review.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2020 Jan 9;1(1):CD011535. doi: 10.1002/14651858.CD011535.pub3. Cochrane Database Syst Rev. 2020. Update in: Cochrane Database Syst Rev. 2021 Apr 19;4:CD011535. doi: 10.1002/14651858.CD011535.pub4. PMID: 31917873 Free PMC article. Updated.
-
Systemic treatments for metastatic cutaneous melanoma.Cochrane Database Syst Rev. 2018 Feb 6;2(2):CD011123. doi: 10.1002/14651858.CD011123.pub2. Cochrane Database Syst Rev. 2018. PMID: 29405038 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
-
Wood Waste Valorization and Classification Approaches: A systematic review.Open Res Eur. 2025 May 6;5:5. doi: 10.12688/openreseurope.18862.2. eCollection 2025. Open Res Eur. 2025. PMID: 40438563 Free PMC article.
References
-
- Elkholi I.E., Robert A., Malouf C., Kuasne H., Drapela S., Macleod G., Hébert S., Pacis A., Calderon V., Kleinman C.L., et al. Targeting the dependence on PIK3C3-mTORC1 signaling in dormancy-prone breast cancer cells blunts metastasis initiation. Cancer Res. 2025;85:2179–2198. doi: 10.1158/0008-5472.CAN-23-2654. - DOI - PMC - PubMed
-
- Goddard E.T., Linde M.H., Srivastava S., Klug G., Shabaneh T.B., Iannone S., Grzelak C.A., Marsh S., Riggio A.I., Shor R.E., et al. Immune evasion of dormant disseminated tumor cells is due to their scarcity and can be overcome by T cell immunotherapies. Cancer Cell. 2024;42:119–134.e112. doi: 10.1016/j.ccell.2023.12.011. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
