

From Antiretroviral to Antibacterial: Deep-Learning-Accelerated Repurposing and In Vitro Validation of Efavirenz Against Gram-Positive Bacteria
- PMID: 40733192
- PMCID: PMC12299921
- DOI: 10.3390/molecules30142925
From Antiretroviral to Antibacterial: Deep-Learning-Accelerated Repurposing and In Vitro Validation of Efavirenz Against Gram-Positive Bacteria
Abstract
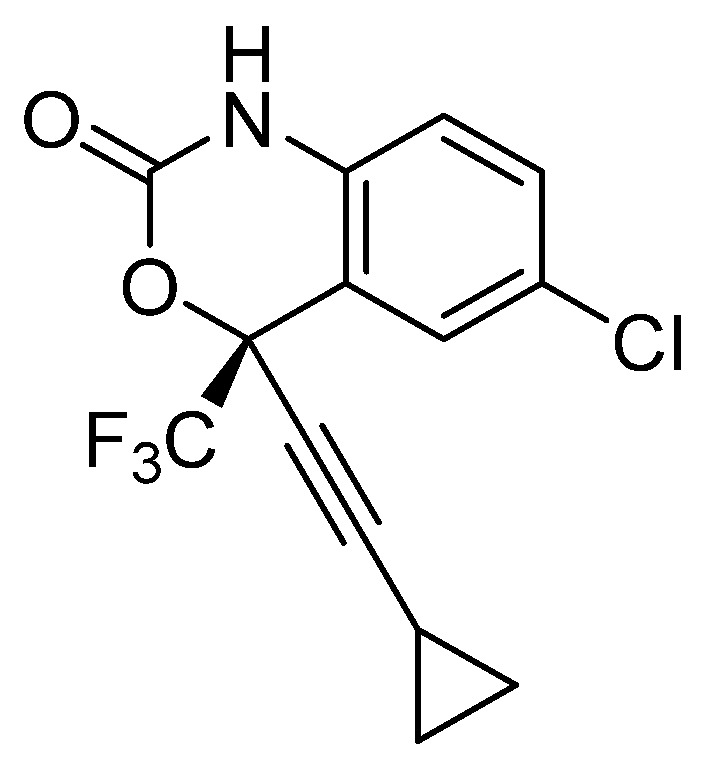
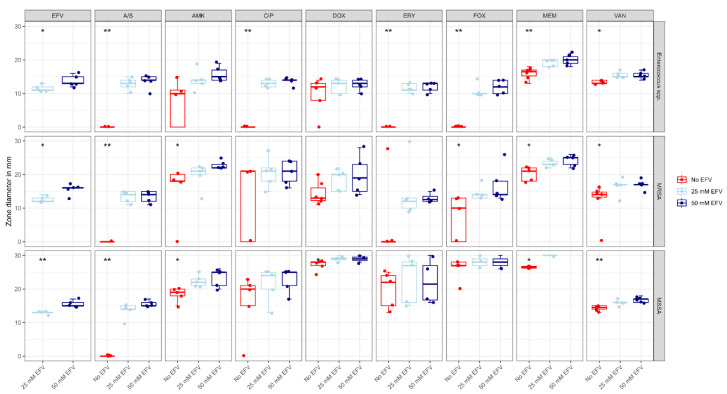
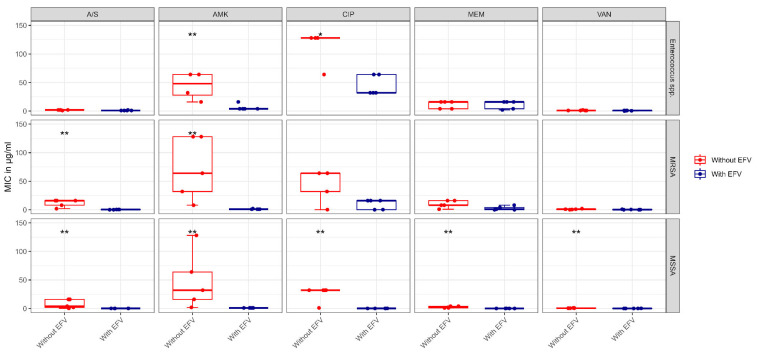
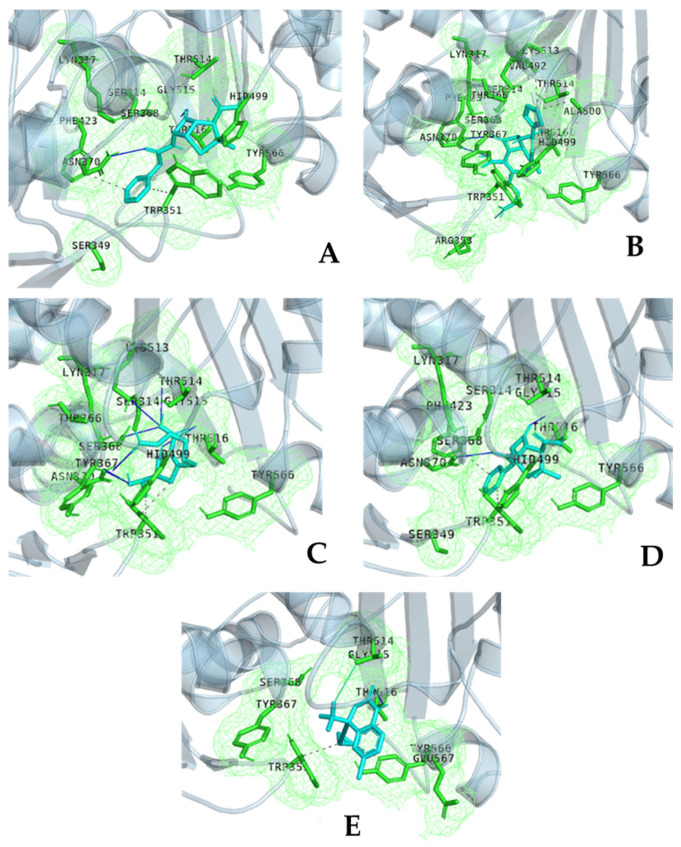
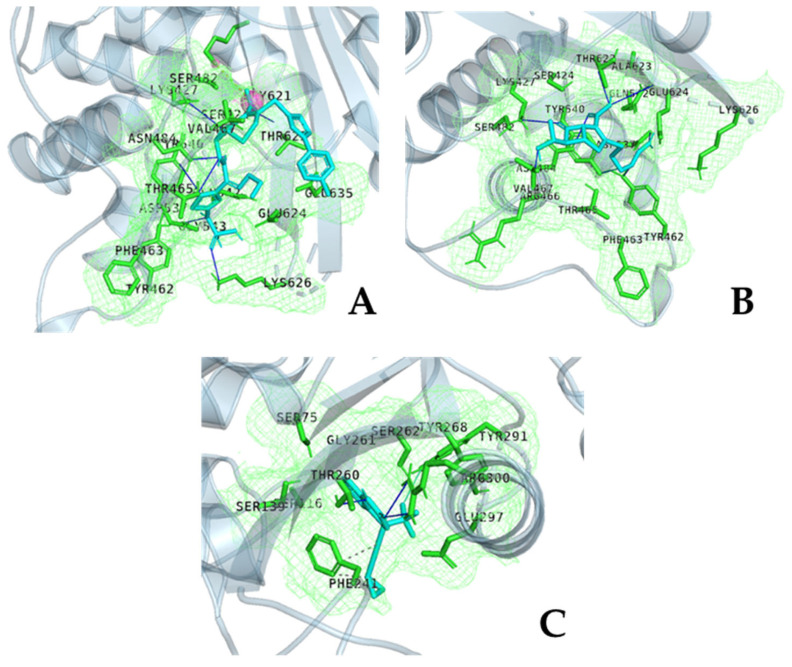
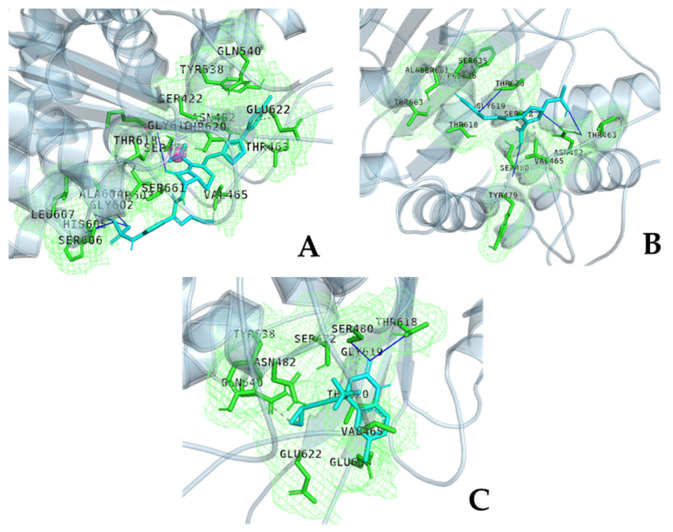
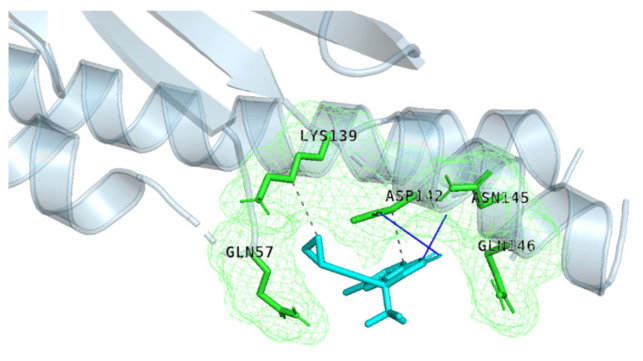
The repurposing potential of Efavirenz (EFV), a clinically established non-nucleoside reverse transcriptase inhibitor, was comprehensively evaluated for its in vitro antibacterial effect either alone or in combination with other antibacterial agents on several Gram-positive clinical strains showing different antibiotic resistance profiles. The binding potential assessed by an in silico study included Penicillin-binding proteins (PBPs) and WalK membrane kinase. Despite the relatively high minimum inhibitory concentration (MIC) limiting the use of EFV as a single antibacterial agent, it exhibits significant synergistic activity at sub-MIC levels when paired with various antibiotics against Enterococcus species and Staphylococcus aureus. EFV showed restored sensitivity of β-lactams against Methicillin-resistant S. aureus (MRSA). It increased the effectiveness of antibiotics tested against Methicillin-sensitive S. aureus (MSSA). It also helped to overcome the intrinsic resistance barrier for several antibiotics in Enterococcus spp. In silico binding studies aligned remarkably with experimental antimicrobial testing results and highlighted the potential of EFV to direct the engagement of PBPs with moderate to strong binding affinities (pKa 5.2-6.1). The dual-site PBP2 binding mechanism emerged as a novel inhibition strategy, potentially circumventing resistance mutations. Special attention should be paid to WalK binding predictions (pKa = 4.94), referring to the potential of EFV to interfere with essential regulatory pathways controlling cell wall metabolism and virulence factor expression. These findings, in general, suggest the possibility of EFV as a promising lead for the development of new antibacterial agents.
Keywords: Efavirenz; antibacterial activity; drug repurposing; in silico binding study.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
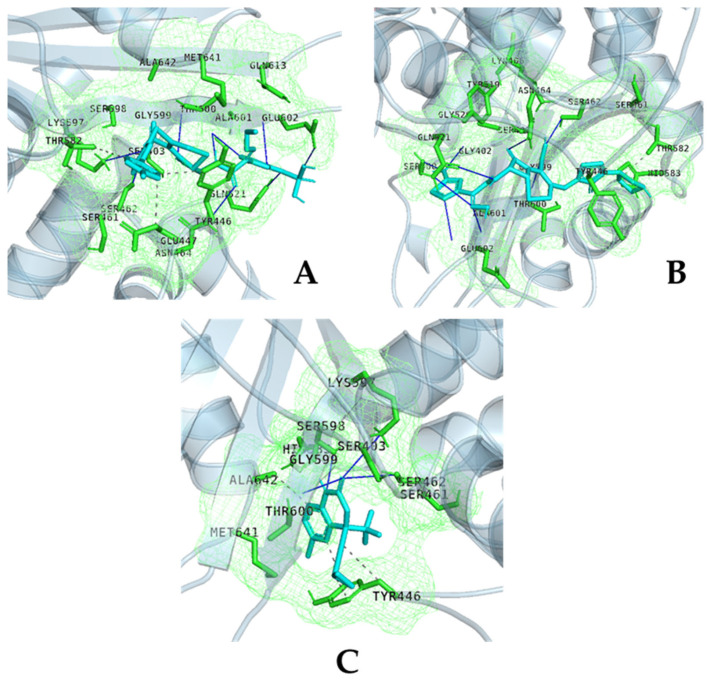
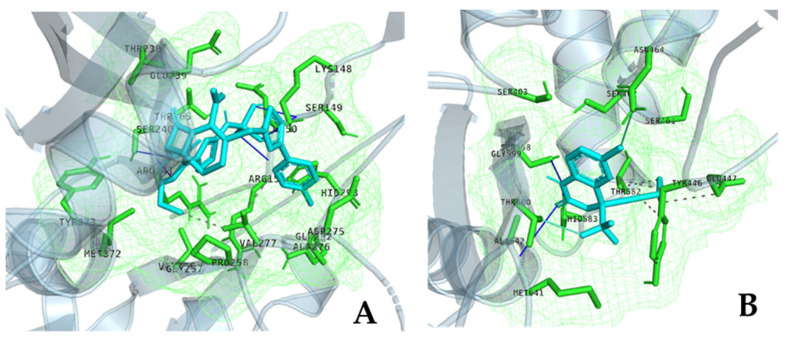
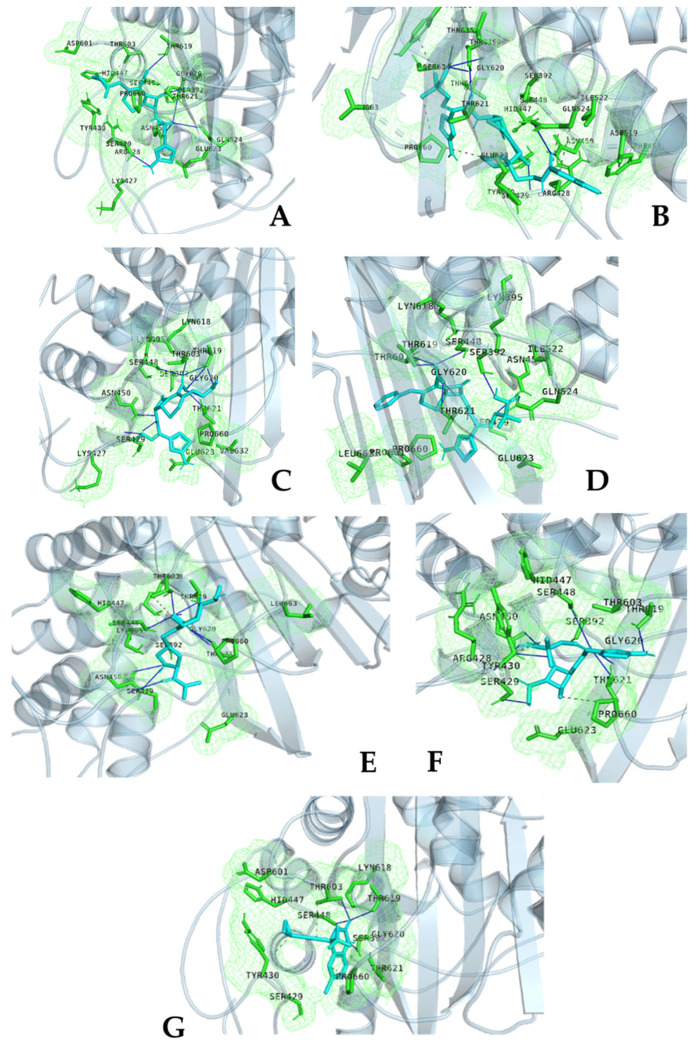
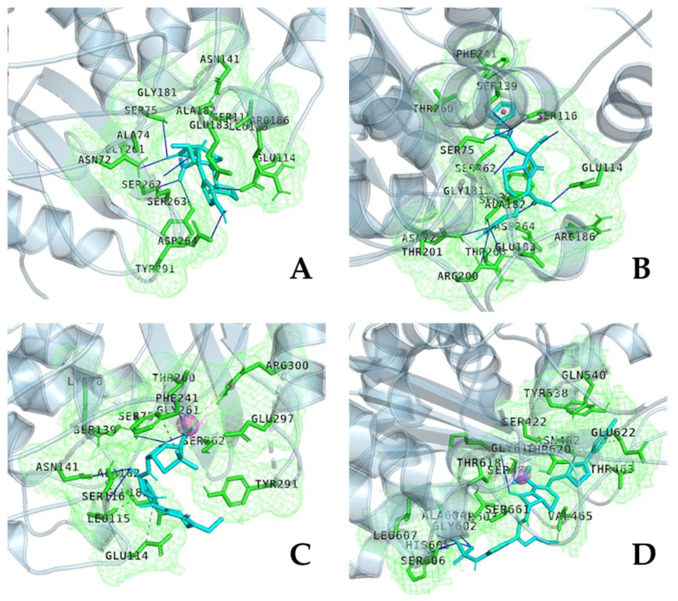
References
-
- World Health Organization . 2019 Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline. World Health Organization; Geneva, Switzerland: 2020.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
