

doi: 10.1016/j.gendis.2025.101546.
eCollection 2025 Nov.
The same genomic variants in the first three exons of KANSL1 can be either benign or causative of Koolen-de Vries syndrome: Definition of a validation procedure
Affiliations
- PMID: 40735694
- PMCID: PMC12304670
- DOI: 10.1016/j.gendis.2025.101546
Item in Clipboard
The same genomic variants in the first three exons of KANSL1 can be either benign or causative of Koolen-de Vries syndrome: Definition of a validation procedure
Genes Dis.
.
No abstract available
Conflict of interest statement
The authors declared no conflict of interests.
Figures
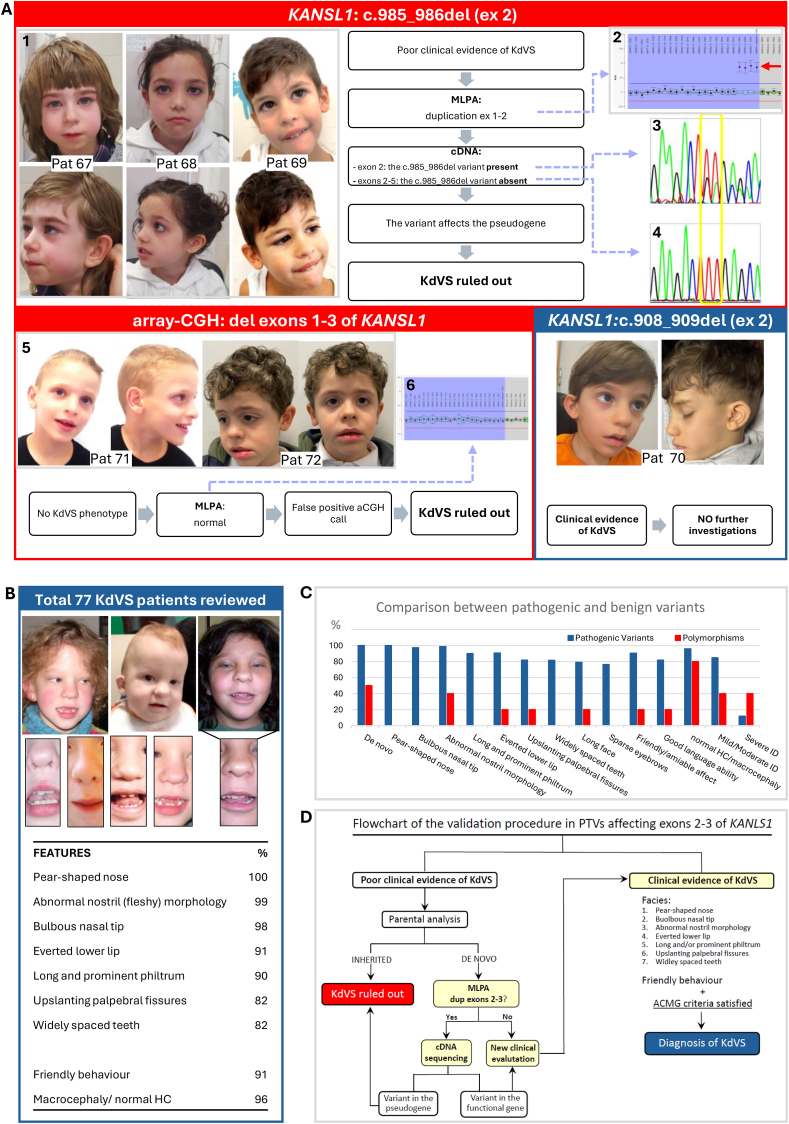
Schematic representation of the clinical and molecular findings that were considered for assessing the pathogenicity of variants in exons 1–3 of KANSL1.(A) Top red panel: Clinical pictures and molecular data of patients 67, 68, and 69 with the c.985_986del variant on exon 2 of KANSL1. (A1) Frontal and lateral view of patients: except for mild upslanting of the palpebral fissures and prominent and long philtrum in patient 67, and of bulbous nasal tip in patient 68, all of them lack the full spectrum of the most distinctive facial features of KdVS. (A2) MLPA analysis: duplication of the first two exons of KANSL1 was observed in all of them. (A3) cDNA sequencing with primers amplifying only exon 2: the c.985_986del variant diagnosed on genomic DNA was detected on cDNA as well. (A4) cDNA sequencing with amplicons spanning exons 2–6: the c.985_986del variant is absent. (A) Bottom red panel: clinical pictures and molecular data of patients 71 and 72 with exons 1–3 deletion called by array-CGH. (A5) Frontal and lateral view of patients: the typical features of KdVS are not appreciable in either. In patient 71, please note the presence of a high forehead; receding hairline on the forehead; large and deep-set eyes; large, medially flaring and sparse eyebrows; high nasal bridge with prominent columella; open mouth, pointed triangular chin and linearized mandibular bones. The clinical hypothesis of a mild presentation of Mowat-Wilson syndrome was confirmed by the detection of the c.3171_3172del variant in exon 10 of ZEB2 (Suppl.P&M). (A6) MLPA analysis: no deletion of exons 1–3 was detected in either. This discrepancy was attributed to a false positive call in array-CGH analysis, contingent on the control samples used. (A) Bottom blue panel: clinical pictures of patient 70 with the de novo c.908_909del in exon 2 of KANSL1. (A6) Frontal and lateral view of the patient: the full spectrum of the most distinctive facial features of KdVS can be appreciated, including the typical nose and mouth conformation. No further molecular investigations were deemed necessary. (B) Revision of 77 KdVS patients (Table S1). Included in the present revision were both patients of personal observation and patients reported in the literature with detailed clinical information and pictures. The facial characteristics of some subjects (No. 26, 28, and 2 previously reported) and the specific nose, mouth, and teeth conformation are highlighted. Below the clinical pictures, a selected list of highly distinctive clinical signs in KdVS is shown, according to their frequency and specificity. (HC=head circumference) (C) Comparison between pathogenic and benign variants in KANSL1. The most frequent clinical manifestations resulting from the revision of 77 KdVS subjects (blue bars) are compared with those of our patients with benign variants in KANSL1 (red bars). The additional features considered in this comparison, in particular sparse eyebrows, long face, good language ability, and the degree of intellectual disability, were not included among the most distinctive clinical signs shown in (B), since most of them are age-related. Please note that the de novo occurrence of variants is not sufficient in assessing their pathogenicity. (ID=intellectual disability) (D) Flowchart of the validation procedure in PTVs affecting exons 2 and 3 of KANSL1. The first step is clinical evaluation (or re-evaluation) of patients: whether the typical KdVS facial features, especially if associated with friendly behavior, can be appreciated, and the ACMG criteria for pathogenicity are satisfied, a definitive diagnosis of KdVS can be made without further molecular investigations (right side). In the absence of the full spectrum of the facial characteristics of KdVS, the inheritance of the detected variant has to be assessed by parental analysis. If the variant is inherited from a non-mosaic healthy parent, it is considered benign, and the diagnosis of KdVS can be ruled out. In cases of de novo PTVs, MLPA analysis of KANSL1 is indicated. If a polymorphic duplication of exons 1–3 of the gene is detected, cDNA sequencing of different amplicons, as illustrated in (A3) and (A4), is indicated, to assess the localization of the variant within either the functional copy of the gene or within its nonfunctional duplicated segment. Whether, by MLPA, a duplication polymorphism in KANSL1 is not detected, and whether, by cDNA sequencing of amplicons spanning from exon 2 to other exons not involved in the duplication polymorphism, the variant can be inferred to affect the functional gene, the expected clinical phenotype is KdVS, and a conscious clinical re-evaluation is recommended, following the here highlighted stringent clinical criteria. Deletions of exons 1–3 of KANSL1 are not considered in the present flowchart. Actually, to date they have never been described in typical KdVS subjects, leading to the conclusion that they are false positive calls when detected in array-CGH analysis, most likely. However, KANSL1 MLPA analysis can be performed to support this conclusion.
References
-
- Zollino M., Marangi G., Ponzi E., et al. Intragenic KANSL1 mutations and chromosome 17q21.31 deletions: broadening the clinical spectrum and genotype-phenotype correlations in a large cohort of patients. J Med Genet. 2015;52(12):804–814. - PubMed
LinkOut - more resources
Full Text Sources
