

Long-read RNA-sequencing reveals transcript-specific regulation in human-derived cortical neurons
- PMID: 40735840
- PMCID: PMC12308531
- DOI: 10.1098/rsob.250200
Long-read RNA-sequencing reveals transcript-specific regulation in human-derived cortical neurons
Abstract
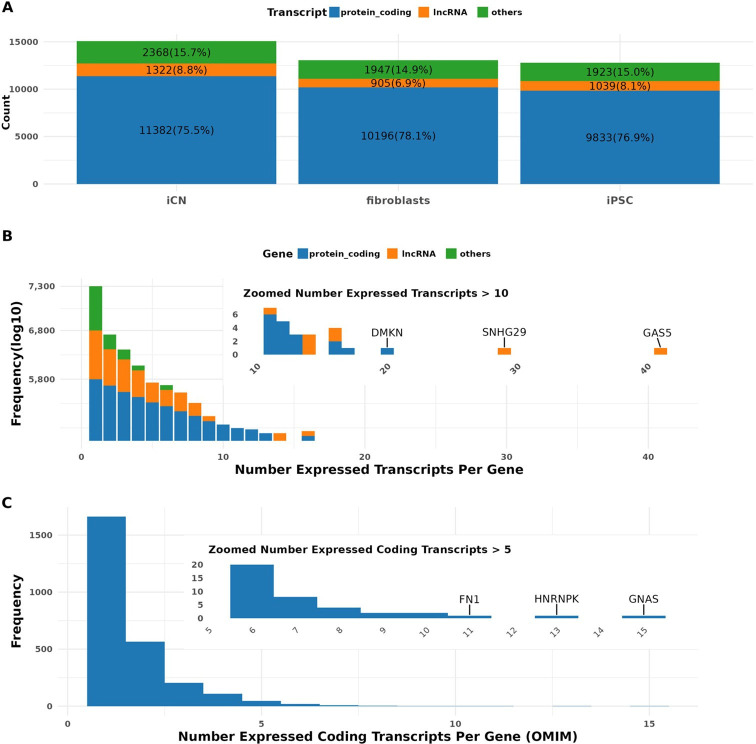
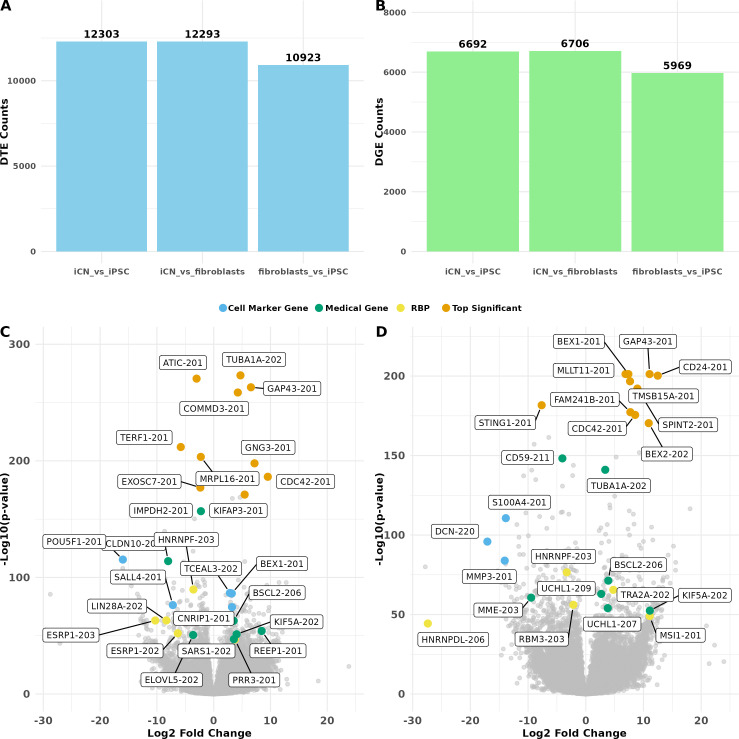
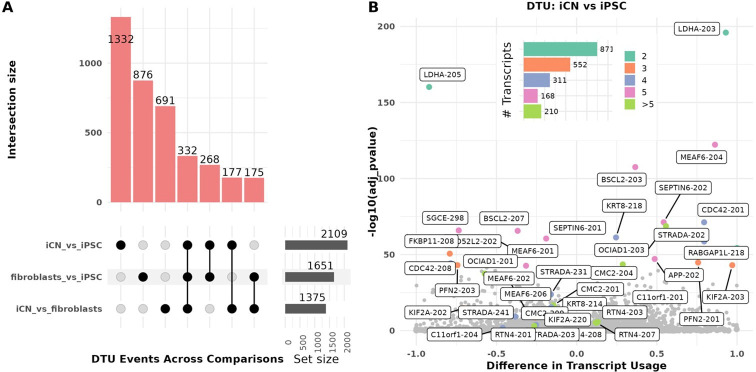
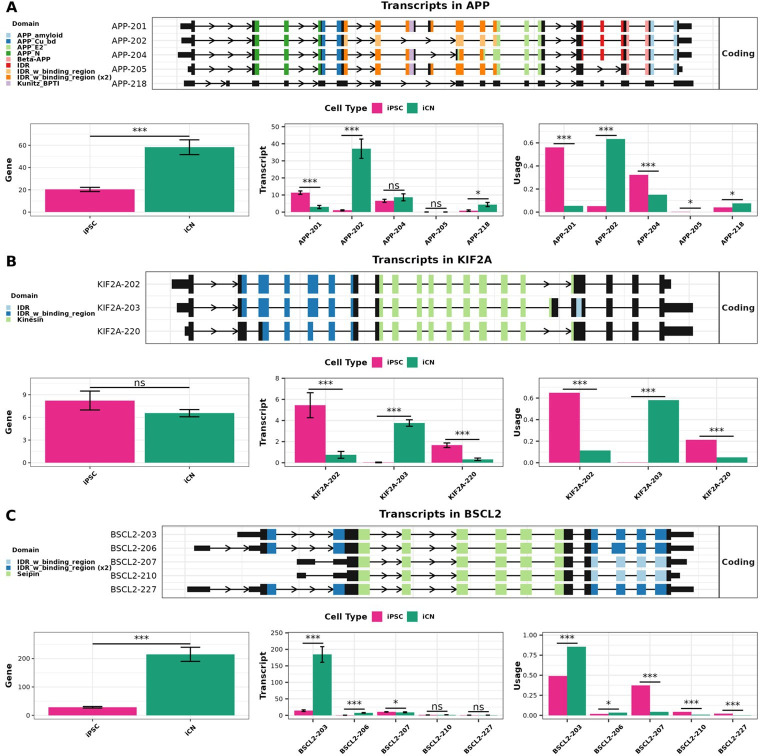
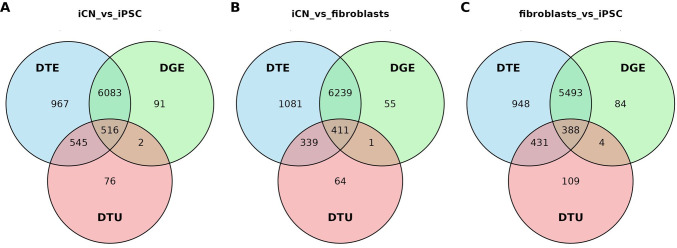
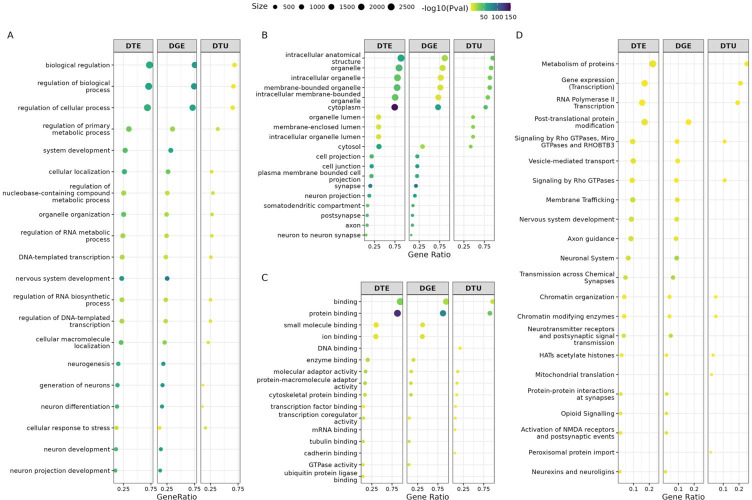
Long-read RNA sequencing has transformed transcriptome analysis by enabling comprehensive mapping of full-length transcripts, providing an unprecedented resolution of transcript diversity, alternative splicing and transcript-specific regulation. In this study, we employed nanopore long-read RNA sequencing to profile the transcriptomes of three cell types commonly used to model brain disorders, human fibroblasts, induced pluripotent stem cells and stem cell-derived cortical neurons, identifying extensive transcript diversity with 15 072 transcripts in stem cell-derived cortical neurons, 13 048 in fibroblasts and 12 759 in induced pluripotent stem cells. Our analyses uncovered 35 519 differential transcript expression events and 5135 differential transcript usage events, underscoring the complexity of transcriptomic regulation across these cell types. Importantly, by integrating differential transcript expression and usage analyses, we gained deeper insights into transcript dynamics that are not captured by gene-level expression analysis alone. Differential transcript usage analysis highlighted transcript-specific changes in disease-relevant genes such as APP, KIF2A and BSCL2, associated with Alzheimer's disease, neuronal migration disorders and degenerative axonopathies, respectively. This added resolution emphasizes the significance of transcript-level variations that often remain hidden in traditional differential gene expression analyses. Overall, our work provides a framework for understanding transcript diversity in both pluripotent and specialized cell types, which can be used to investigate transcriptomic changes in disease states in future work. Additionally, this study underscores the utility of differential transcript usage analysis in advancing our understanding of neurodevelopmental and neurodegenerative diseases, paving the way for identifying transcript-specific therapeutic targets.
Keywords: alternative splicing; human-derived cortical neurons; induced pluripotent stem cells; long-read RNA-sequencing; transcript usage; transcriptomics.
Conflict of interest statement
We declare we have no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources