

Mouse lemur cell atlas informs primate genes, physiology and disease
- PMID: 40739355
- PMCID: PMC12328237
- DOI: 10.1038/s41586-025-09114-8
Mouse lemur cell atlas informs primate genes, physiology and disease
Abstract
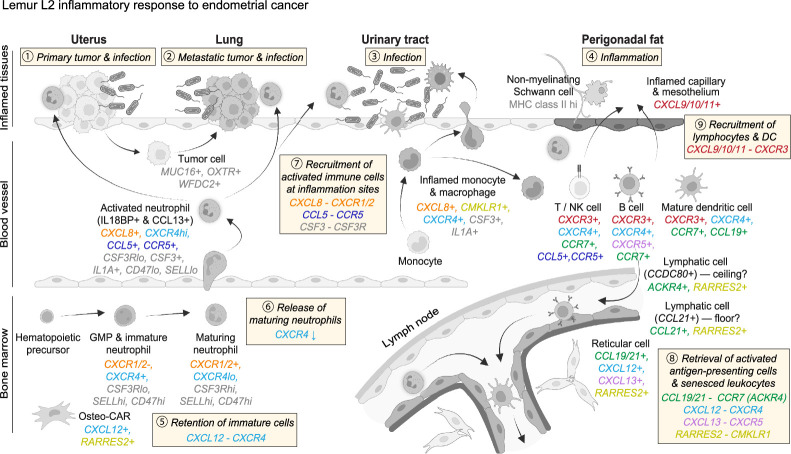
Mouse lemurs (Microcebus spp.) are an emerging primate model organism, but their genetics, cellular and molecular biology remain largely unexplored. In an accompanying paper1, we performed large-scale single-cell RNA sequencing of 27 organs from mouse lemurs. We identified more than 750 molecular cell types, characterized their transcriptomic profiles and provided insight into primate evolution of cell types. Here we use the generated atlas to characterize mouse lemur genes, physiology, disease and mutations. We uncover thousands of previously unidentified lemur genes and hundreds of thousands of new splice junctions including over 85,000 primate splice junctions missing in mice. We systematically explore the lemur immune system by comparing global expression profiles of key immune genes in health and disease, and by mapping immune cell development, trafficking and activation. We characterize primate-specific and lemur-specific physiology and disease, including molecular features of the immune program, lemur adipocytes and metastatic endometrial cancer that resembles the human malignancy. We present expression patterns of more than 400 primate genes missing in mice, many with similar expression patterns to humans and some implicated in human disease. Finally, we provide an experimental framework for reverse genetic analysis by identifying naturally occurring nonsense mutations in three primate immune genes missing in mice and by analysing their transcriptional phenotypes. This work establishes a foundation for molecular and genetic analyses of mouse lemurs and prioritizes primate genes, isoforms, physiology and disease for future study.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures















References
-
- Terrien, J. et al. Metabolic and genomic adaptations to winter fattening in a primate species, the grey mouse lemur (Microcebus murinus). Int. J. Obesity42, 221–230 (2018). - PubMed
-
- Bons, N., Rieger, F., Prudhomme, D., Fisher, A. & Krause, K.-H. Microcebus murinus: a useful primate model for human cerebral aging and Alzheimer’s disease? Genes Brain Behav.5, 120–130 (2006). - PubMed
MeSH terms
Grants and funding
- R35 GM136433/GM/NIGMS NIH HHS/United States
- P30 EY026877/EY/NEI NIH HHS/United States
- R25 CA180993/CA/NCI NIH HHS/United States
- P01 AG036695/AG/NIA NIH HHS/United States
- R35 GM139517/GM/NIGMS NIH HHS/United States
- K99 AG066963/AG/NIA NIH HHS/United States
- R01 AR073248/AR/NIAMS NIH HHS/United States
- R01 NS050835/NS/NINDS NIH HHS/United States
- P30 DK116074/DK/NIDDK NIH HHS/United States
- R01 AG068667/AG/NIA NIH HHS/United States
- R01 DC016892/DC/NIDCD NIH HHS/United States
- T32 AI007290/AI/NIAID NIH HHS/United States
- R01 GM061986/GM/NIGMS NIH HHS/United States
- DP2 AI138242/AI/NIAID NIH HHS/United States
- R01 AI024258/AI/NIAID NIH HHS/United States
- R01 GM116847/GM/NIGMS NIH HHS/United States
- T15 LM007033/LM/NLM NIH HHS/United States
- RF1 AG077443/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
