

Whole Exome Sequencing Identified a Stop-Gained Mutation in DYSF Gene Associated With Dysferlinopathy in an Iranian Family
- PMID: 40740503
- PMCID: PMC12310322
- DOI: 10.1155/ijog/9103068
Whole Exome Sequencing Identified a Stop-Gained Mutation in DYSF Gene Associated With Dysferlinopathy in an Iranian Family
Abstract
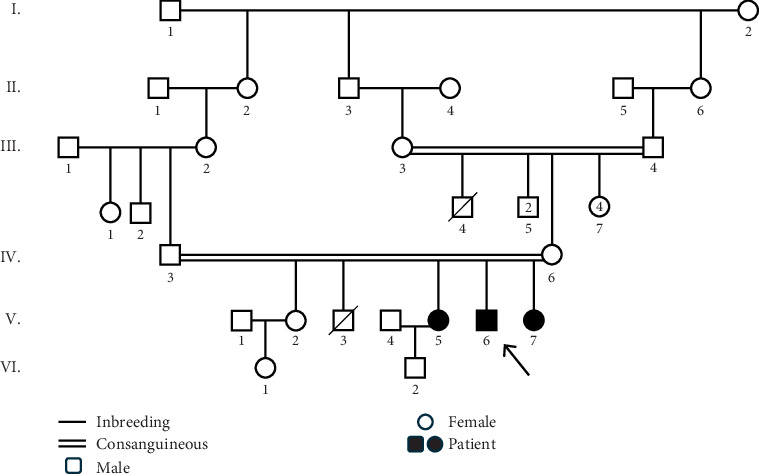
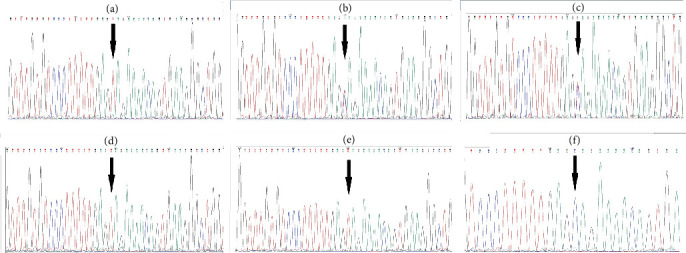
Introduction: Muscular dystrophy (MD) refers to a group of hereditary disorders characterized by progressive muscle degeneration, often caused by a deficiency or insufficient levels of glycoproteins in muscle cell membranes. Mutations in various genes lead to different types of MD, each with distinct clinical manifestations and inheritance patterns. The genetic heterogeneity of MD complicates the identification of the causative genes. Materials and Methods: This research was conducted to identify the genetic basis of MD in an Iranian family with three affected members. Whole exome sequencing (WES) was performed on a proband who had initially been misdiagnosed with polymyositis. Following the identification of the disease-causing variant via WES, cosegregation analysis was carried out among two affected siblings, the asymptomatic parents, and one unaffected sibling. Results: WES identified a homozygous nonsense variant (c.6001C>T, p.Gln2001Ter) in Exon 53 of the DYSF gene, which encodes dysferlin, a transmembrane protein essential for membrane protection and repair following damage. This stop-gain mutation results in a nonfunctional truncated protein lacking the transmembrane helix, preventing its anchorage to the membrane. Dysfunction of dysferlin is associated with limb-girdle muscular dystrophy 2B (LGMD2B) and Miyoshi myopathy. Discussion: Bioinformatics analyses and clinical findings confirmed the pathogenicity of this variant in a homozygous state, consistent with autosomal recessive inheritance. Furthermore, structural modeling suggested that the mutation significantly disrupts the tertiary structure of dysferlin. Since the disorder onset in the proband and his two affected sisters began in the proximal limb muscles, the condition was classified as LGMD. The study highlights the diagnostic value of WES in accurately identifying disease-causing variants, offering substantial improvements in time and cost efficiency over conventional diagnostic procedures.
Keywords: DYSF; LGMD; WES; dysferlin; muscular dystrophy.
Copyright © 2025 Saba Baghshomali et al. International Journal of Genomics published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

Similar articles
-
Dysferlinopathy as cause of long-term hyperCKemia with preserved strength.Orphanet J Rare Dis. 2025 Jun 22;20(1):317. doi: 10.1186/s13023-025-03850-w. Orphanet J Rare Dis. 2025. PMID: 40545540 Free PMC article.
-
A novel follicle-stimulating hormone receptor mutation causing primary ovarian failure: a fertility application of whole exome sequencing.Hum Reprod. 2016 Apr;31(4):905-14. doi: 10.1093/humrep/dew025. Epub 2016 Feb 23. Hum Reprod. 2016. PMID: 26911863 Free PMC article.
-
[Clinical and genetic characteristics of familial cases with Glucose transporter 1 deficiency syndrome].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025 Apr 10;42(4):424-432. doi: 10.3760/cma.j.cn511374-20241009-00524. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025. PMID: 40555655 Chinese.
-
Genetic Homogeneity of a TDP1 Variant, c.1478A>G, as the Main Disease-Causing Variant of Spinocerebellar Ataxia With Axonal Neuropathy 1 (SCAN1) in the Middle East: A Systematic Review.Pediatr Neurol. 2025 Mar;164:41-52. doi: 10.1016/j.pediatrneurol.2024.12.011. Epub 2024 Dec 30. Pediatr Neurol. 2025. PMID: 39848142
-
[Volume and health outcomes: evidence from systematic reviews and from evaluation of Italian hospital data].Epidemiol Prev. 2013 Mar-Jun;37(2-3 Suppl 2):1-100. Epidemiol Prev. 2013. PMID: 23851286 Italian.
References
LinkOut - more resources
Full Text Sources
