

A side-by-side comparison of variant function measurements using deep mutational scanning and base editing
- PMID: 40744495
- PMCID: PMC12311789
- DOI: 10.1093/nar/gkaf738
A side-by-side comparison of variant function measurements using deep mutational scanning and base editing
Abstract
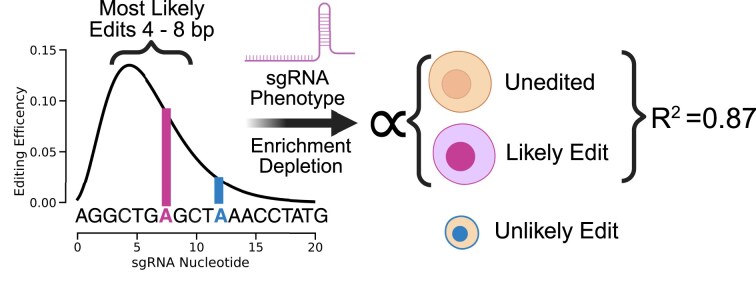
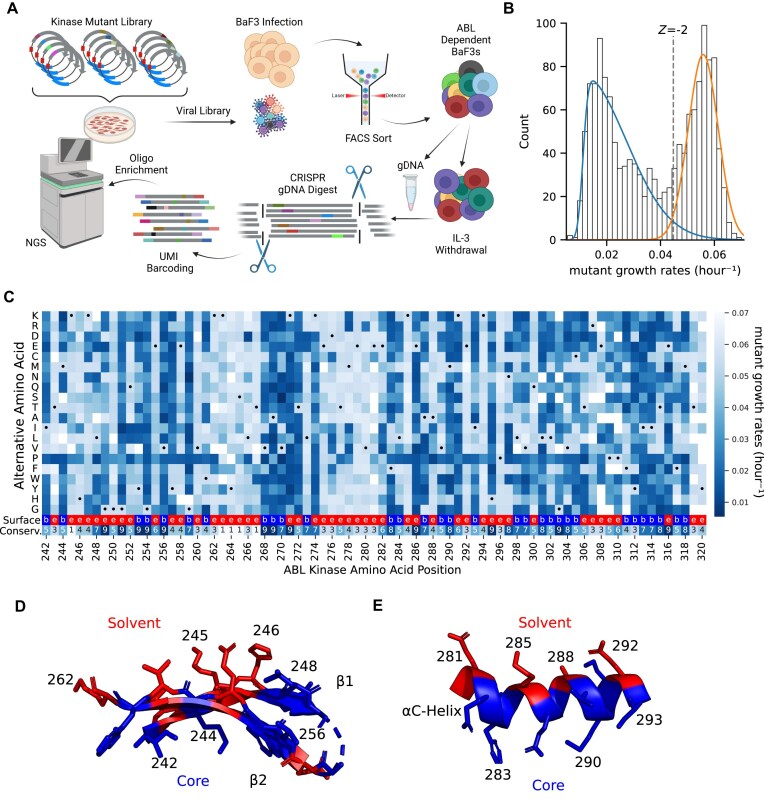
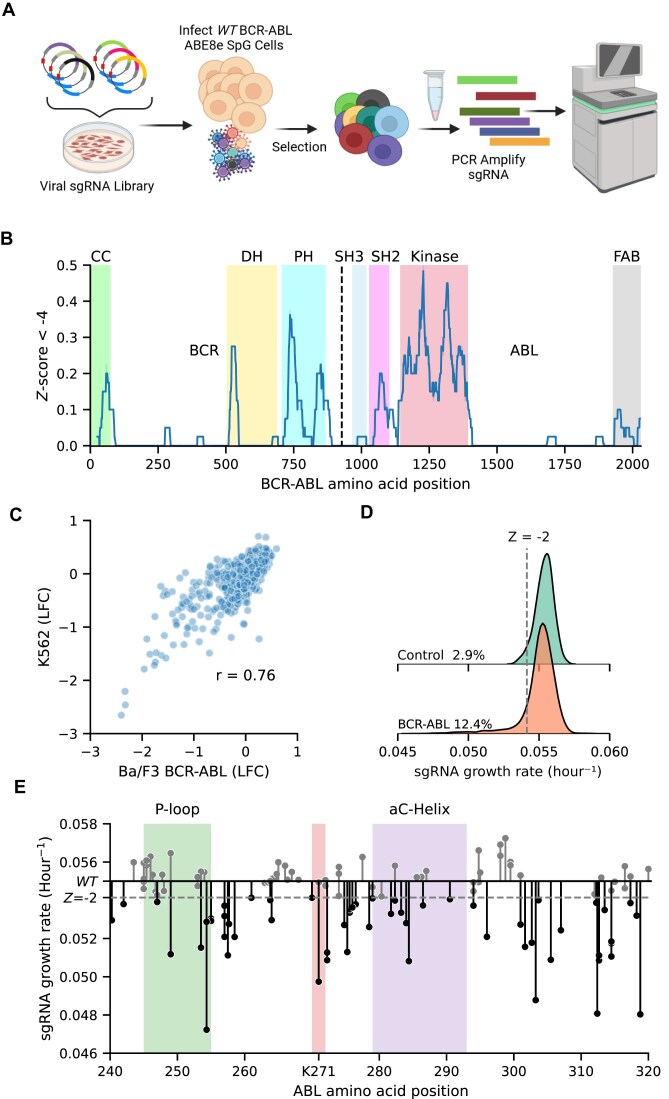
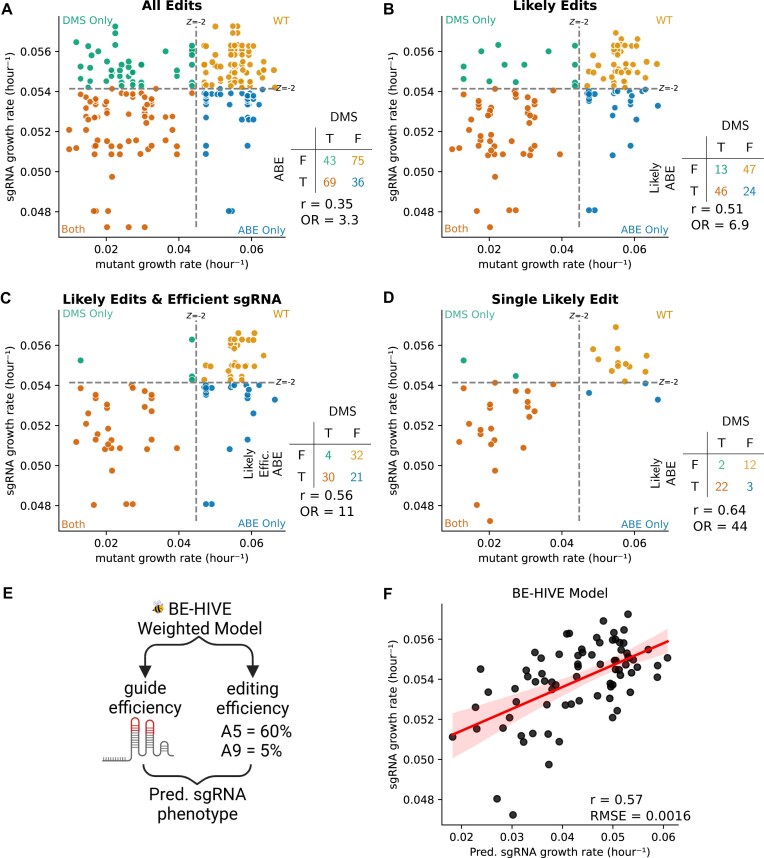
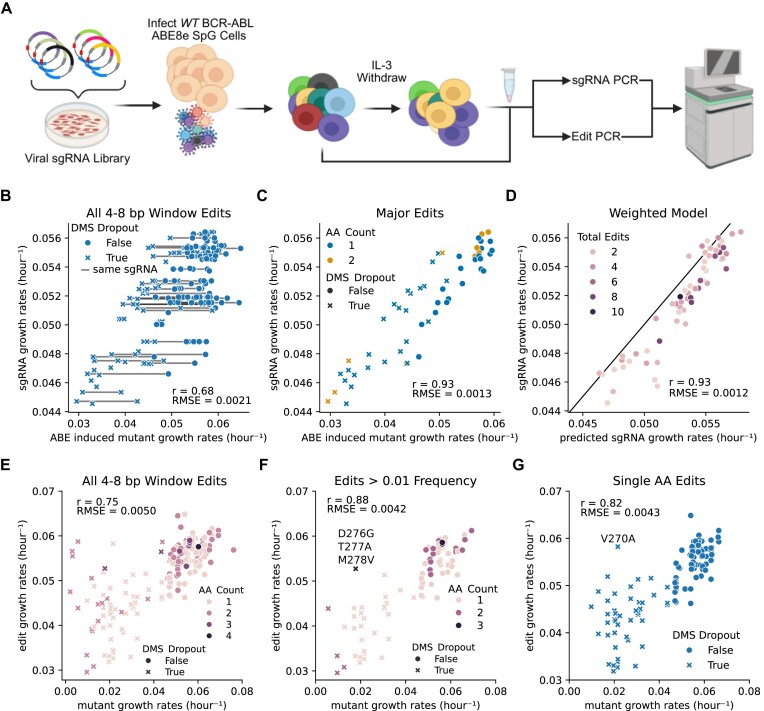
Variant annotation is a crucial objective in mammalian functional genomics. Deep mutational scanning (DMS) using saturation libraries of complementary DNAs (cDNAs) is a well-established method for annotating human gene variants, but CRISPR base editing (BE) is emerging as an alternative. However, questions remain about how well high-throughput BE measurements can annotate variant function and the extent of downstream experimental validation required. This study is the first direct comparison of cDNA DMS and BE in the same lab and cell line. We focus on how well short guide RNA (sgRNA) depletion or enrichment is explained by the predicted edits within the editing "window" defined by the sgRNA. The most likely predicted edits enhance the agreement between a "gold standard" DMS dataset and a BE screen. A simple filter for sgRNAs making single edits in their window could sufficiently annotate a large proportion of variants directly from sgRNA sequencing of large pools. When multi-edit guides are unavoidable, directly measuring edits in medium-sized validation pools can recover high-quality variant annotation data. Our data show a surprisingly high degree of correlation between base editor data and gold standard DMS. We suggest that the main variable measured in base editor screens is the desired base edits.
© The Author(s) 2025. Published by Oxford University Press.
Conflict of interest statement
J.R.P. is a co-founder of RedAce Bio. J.R.P. is a co-founder and consultant for Theseus Pharmaceuticals. J.R.P. received equity from Theseus Pharmaceuticals, MOMA therapeutics and RedAce Bio. J.R.P. has consulted/consults for MOMA therapeutics, Curie.Bio, Third Rock Ventures, Takeda Pharmaceuticals, Galapagos Pharmaceuticals, and Roche/Genentech. J.R.P. has received honoraria and travel expenses from Roche/Genentech, Third Rock Ventures, and Theseus Pharmaceuticals. H.I., M.T., and J.R. are co-founders of Atlas Biotech.
Figures
Update of
-
A side-by-side comparison of variant function measurements using deep mutational scanning and base editing.bioRxiv [Preprint]. 2024 Sep 26:2024.06.30.601444. doi: 10.1101/2024.06.30.601444. bioRxiv. 2024. Update in: Nucleic Acids Res. 2025 Jul 19;53(14):gkaf738. doi: 10.1093/nar/gkaf738. PMID: 39005366 Free PMC article. Updated. Preprint.
Similar articles
-
A side-by-side comparison of variant function measurements using deep mutational scanning and base editing.bioRxiv [Preprint]. 2024 Sep 26:2024.06.30.601444. doi: 10.1101/2024.06.30.601444. bioRxiv. 2024. Update in: Nucleic Acids Res. 2025 Jul 19;53(14):gkaf738. doi: 10.1093/nar/gkaf738. PMID: 39005366 Free PMC article. Updated. Preprint.
-
CRISPR-BEasy: a free web-based service for designing sgRNA tiling libraries for CRISPR-dependent base editing screens.Nucleic Acids Res. 2025 Jul 7;53(W1):W193-W202. doi: 10.1093/nar/gkaf382. Nucleic Acids Res. 2025. PMID: 40377102 Free PMC article.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
A rapid and systematic review of the clinical effectiveness and cost-effectiveness of paclitaxel, docetaxel, gemcitabine and vinorelbine in non-small-cell lung cancer.Health Technol Assess. 2001;5(32):1-195. doi: 10.3310/hta5320. Health Technol Assess. 2001. PMID: 12065068
-
Management of urinary stones by experts in stone disease (ESD 2025).Arch Ital Urol Androl. 2025 Jun 30;97(2):14085. doi: 10.4081/aiua.2025.14085. Epub 2025 Jun 30. Arch Ital Urol Androl. 2025. PMID: 40583613 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
