

A feasibility study of enzymatic methylation sequencing of cell-free DNA from cerebrospinal fluid of pediatric central nervous system tumor patients for molecular classification
- PMID: 40746948
- PMCID: PMC12311925
- DOI: 10.1093/noajnl/vdaf159
A feasibility study of enzymatic methylation sequencing of cell-free DNA from cerebrospinal fluid of pediatric central nervous system tumor patients for molecular classification
Abstract
Background: Array-based DNA methylation profiling is the gold standard for central nervous system (CNS) tumor molecular classification, but requires over 100 ng input DNA from surgical tissue. Cell-free tumor DNA (cfDNA) in cerebrospinal fluid (CSF) offers an alternative for diagnosis and disease monitoring. This study aimed to test the utilization of enzymatic DNA methylation sequencing (EM-seq) methods to overcome input DNA limitations.
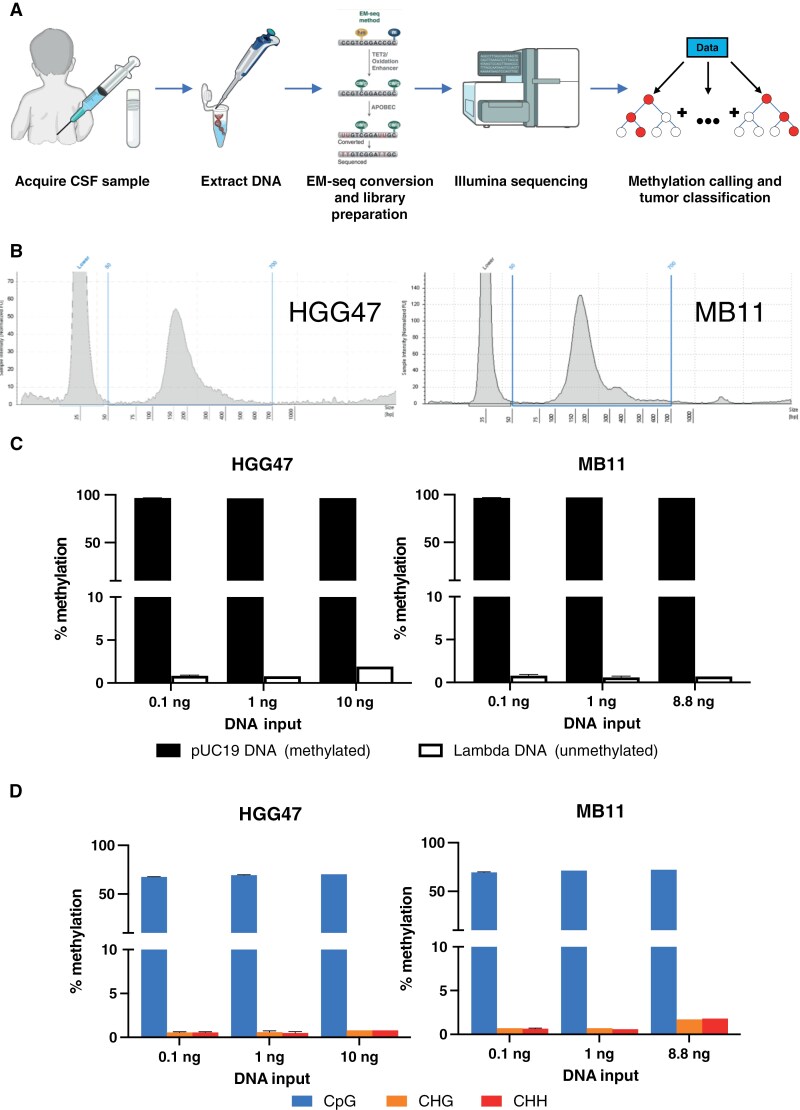
Methods: We used the NEBNext EM-seq v2 kit on various amounts of cfDNA, as low as 0.1 ng, extracted from archival CSF samples of 10 patients with CNS tumors. Tumor classification was performed via MNP-Flex using CpG sites overlapping those on the MethylationEPIC array.
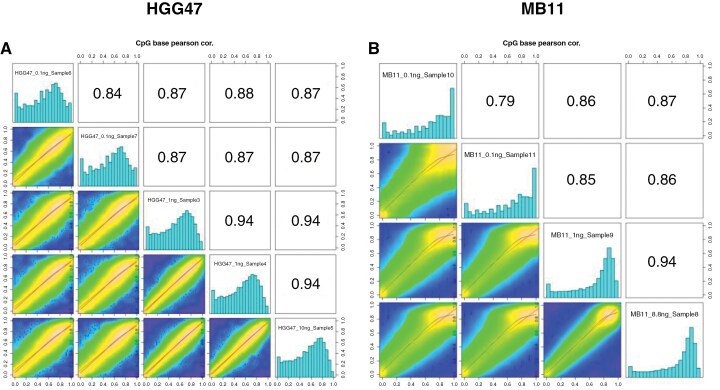
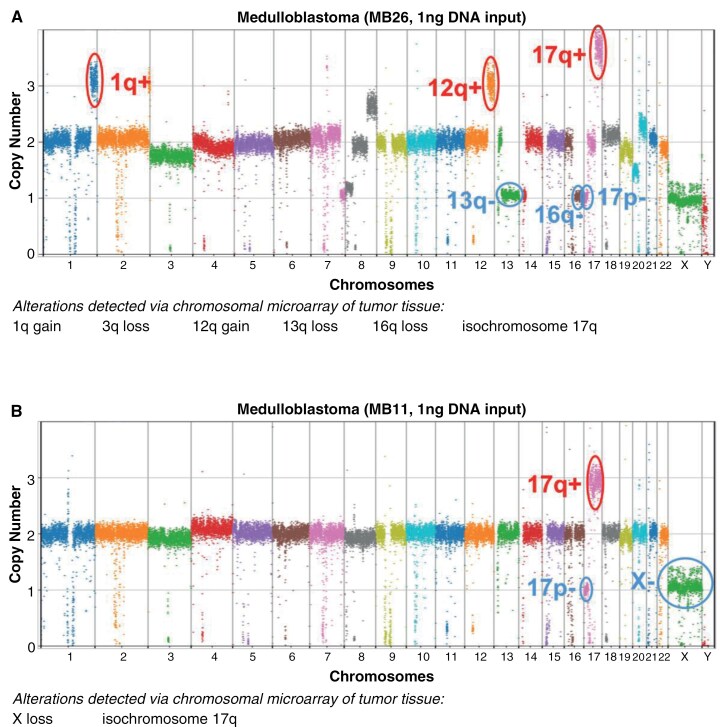
Results: EM-seq provided sufficient genomic coverage for 10 and 1 ng input DNA samples to generate global DNA methylation profiles. Samples with 0.1 ng input showed lower coverage due to read duplication. Methylation levels for CpG sites with at least 5× coverage were highly correlated across various input DNA amounts, indicating that lower input cfDNA can still be used for tumor classification. The MNP-Flex classifier, trained on tissue DNA methylation data, successfully predicted CNS tumor types for 7 out of 10 CSF samples using EM-seq methylation data with only 1 ng of input cfDNA, consistent with diagnoses based on tissue MethylationEPIC classification and/or histopathology. Additionally, we detected focal and arm-level copy number alterations previously identified via clinical cytogenetics of tumor tissue.
Conclusions: This study demonstrated the feasibility of CNS tumor molecular classification based on CSF using the EM-seq approach, and establishes potential sample quality limitations for future studies.
Keywords: CNS tumor classification; MNP-flex; cell-free DNA; enzymatic methylation sequencing; molecular diagnosis.
© The Author(s) 2025. Published by Oxford University Press, the Society for Neuro-Oncology and the European Association of Neuro-Oncology.
Conflict of interest statement
A.M.T., J.T.L., A.P., A.T., J.M., M.J.B., D.S.H., E.C., X.S., P.K.-S.N., and J.J.G.—none declared. F.S.—co-founder and shareholder of Heidelberg Epignostix GmbH. C.C.L.—early access to the NEBNext Enzymatic Methyl-seq v2 Kit was provided by NEB.
Figures
References
-
- Jamal-Hanjani M, Wilson GA, Horswell S, et al. Detection of ubiquitous and heterogeneous mutations in cell-free DNA from patients with early-stage non-small-cell lung cancer. Ann Oncol. 2016;27(5):862–867. - PubMed
LinkOut - more resources
Full Text Sources