

Research progress on mesenchymal stem cell‑derived exosomes in the treatment of osteoporosis induced by knee osteoarthritis (Review)
- PMID: 40747674
- PMCID: PMC12339181
- DOI: 10.3892/ijmm.2025.5601
Research progress on mesenchymal stem cell‑derived exosomes in the treatment of osteoporosis induced by knee osteoarthritis (Review)
Abstract
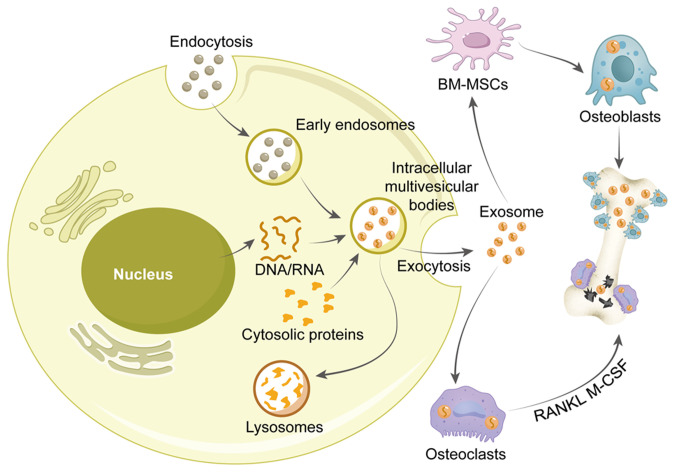
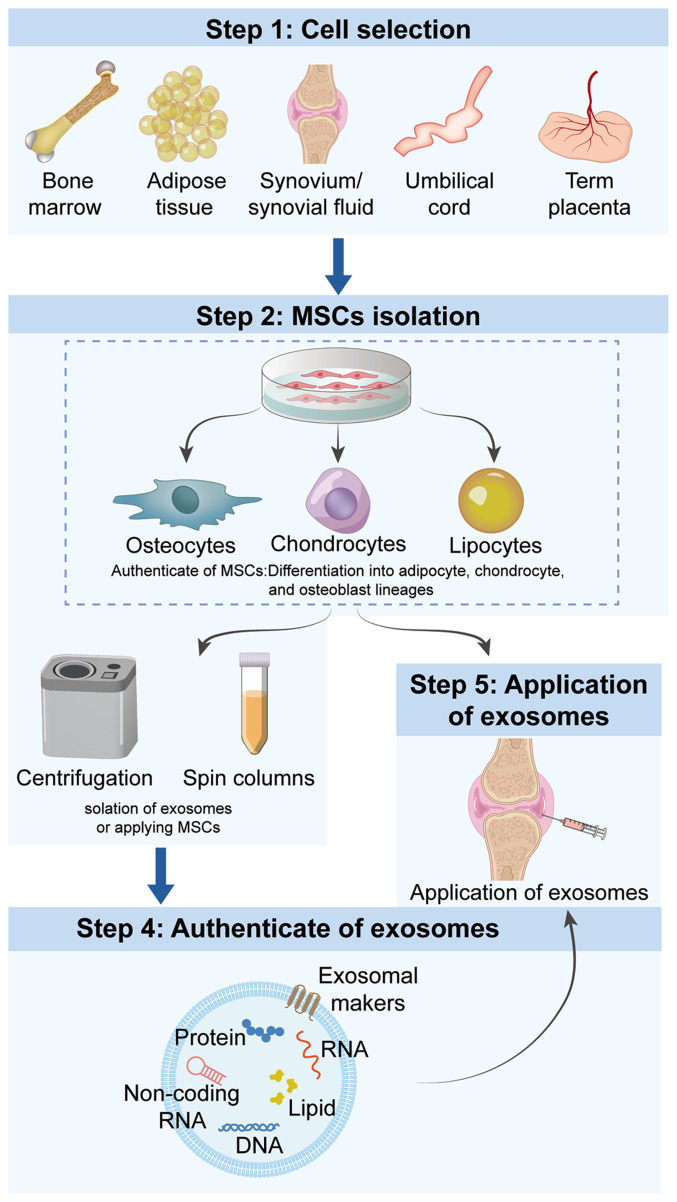
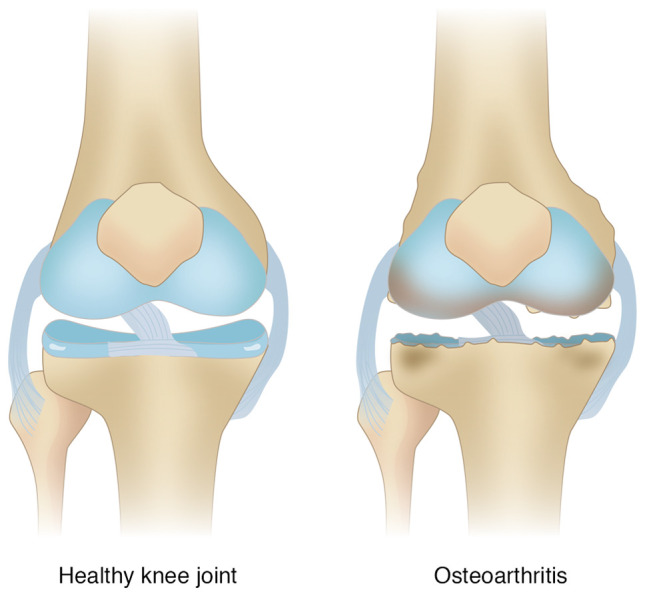
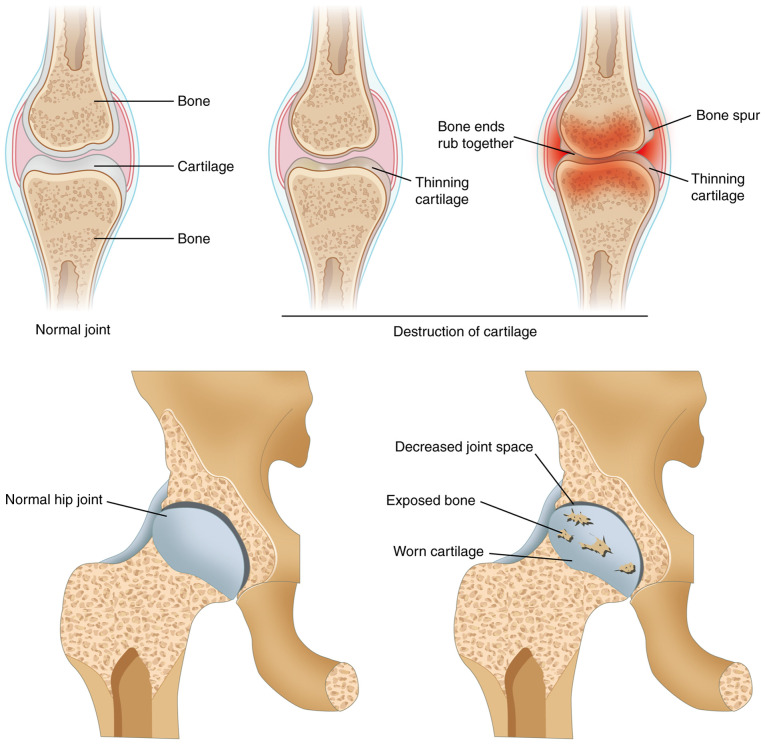
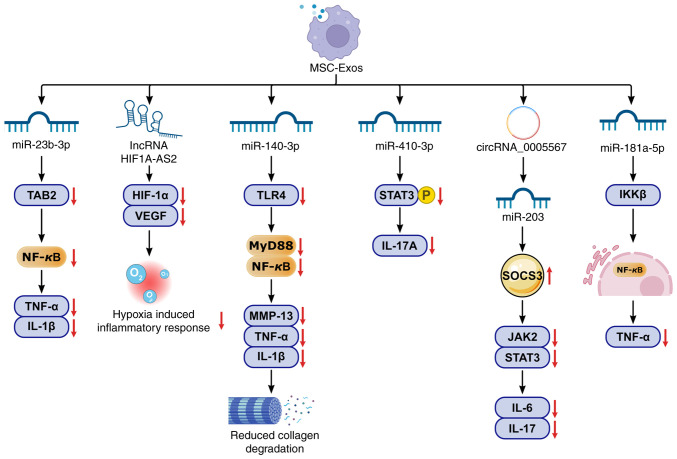
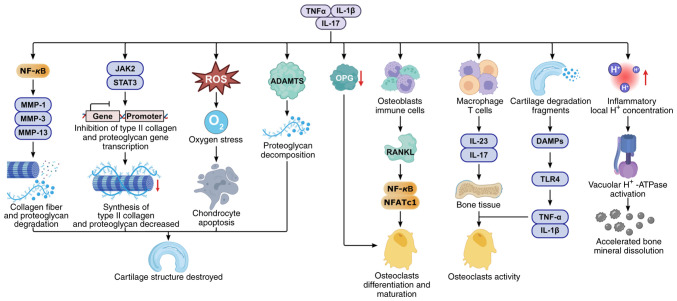
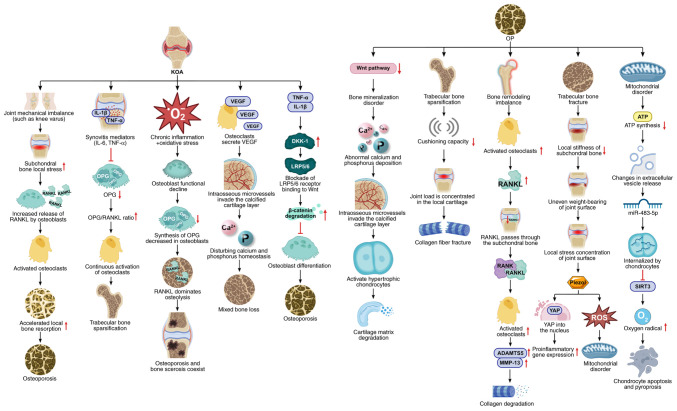
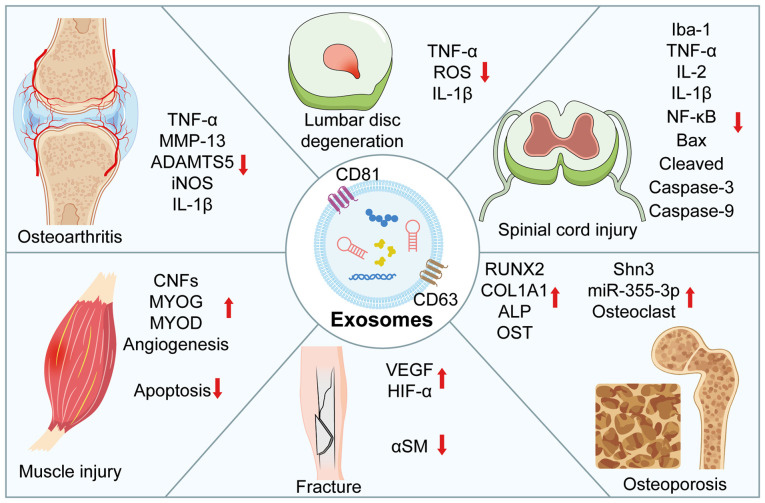
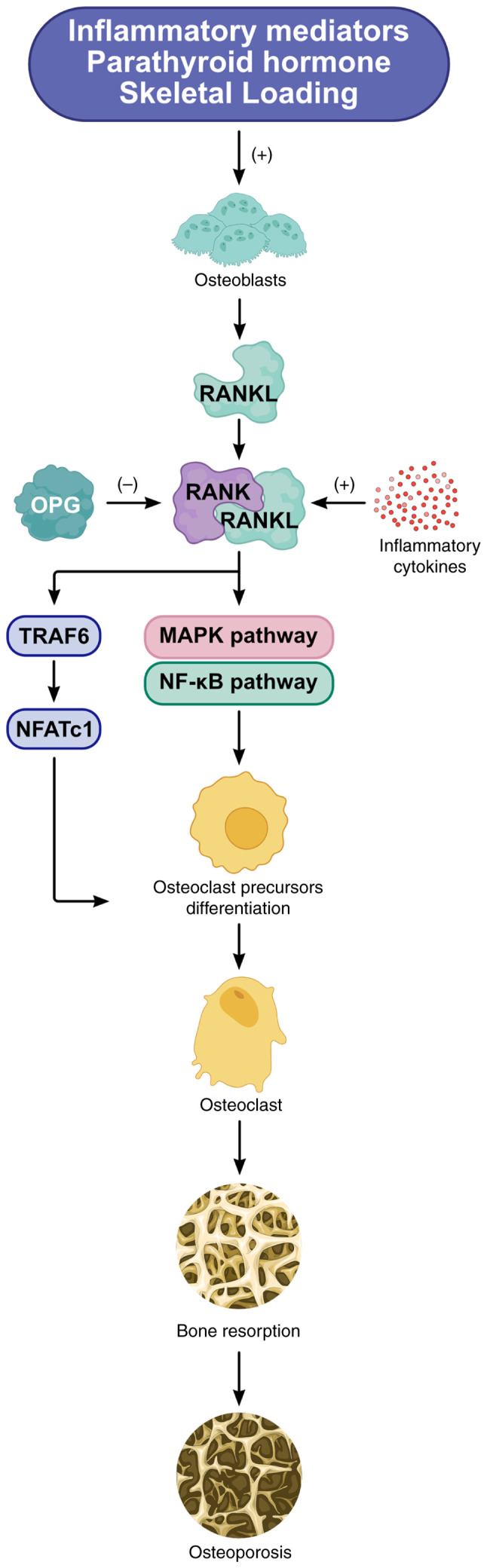
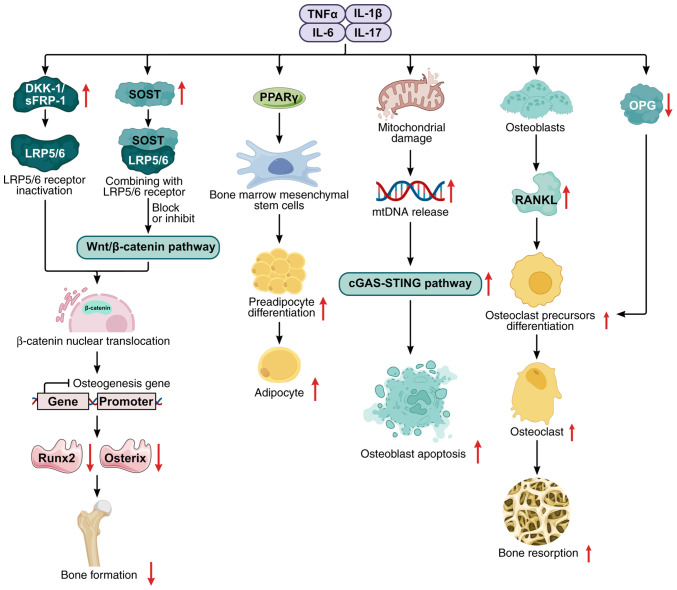
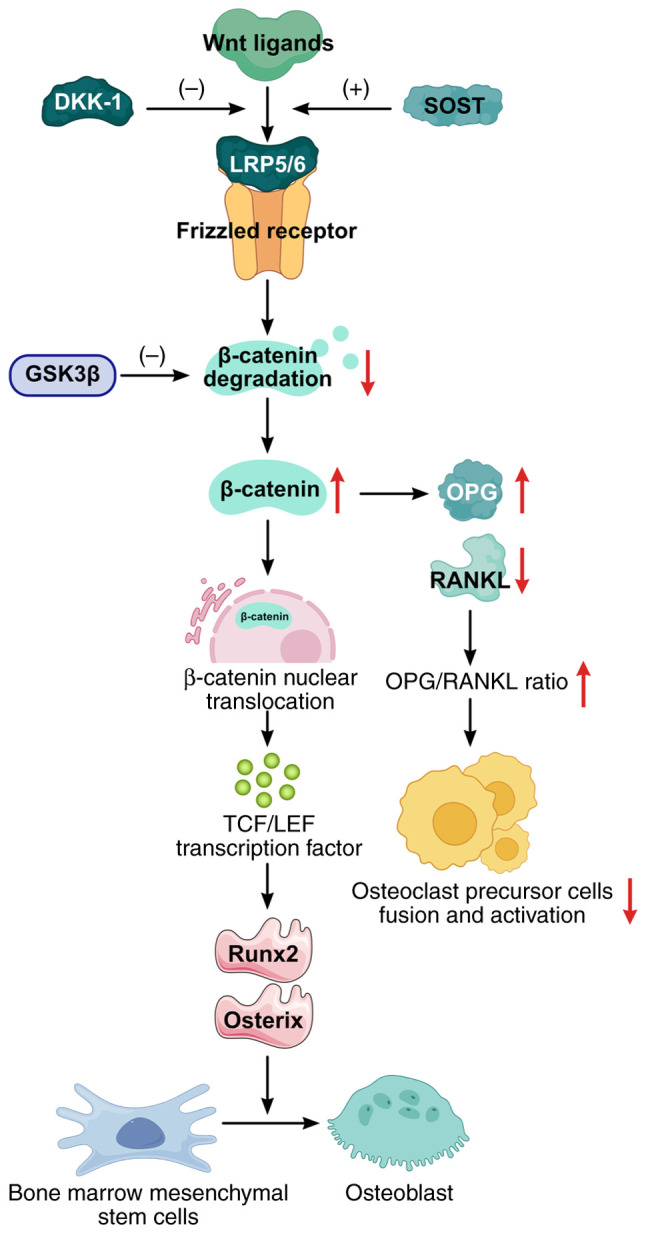
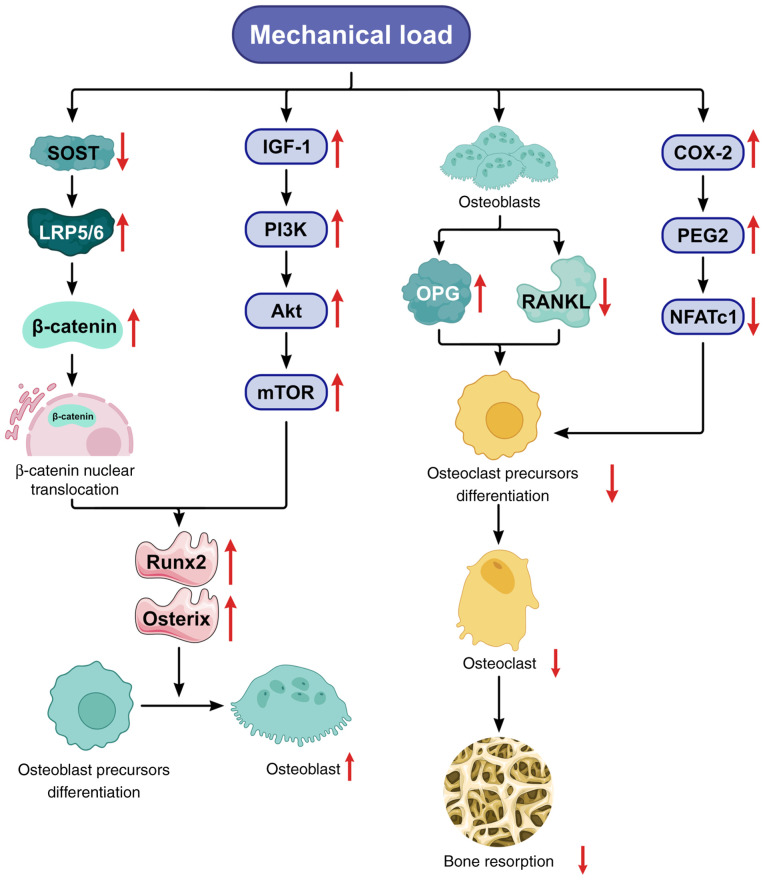
Knee osteoarthritis (KOA) and osteoporosis (OP) are closely related, age‑related, degenerative orthopedic conditions. Elderly patients with OP frequently develop concurrent KOA, with high co‑occurrence rates. Studies indicate that OP significantly increases KOA risk and that these conditions mutually exacerbate each other. Anti‑OP therapies show significant efficacy in KOA management, substantially delaying disease progression. Mesenchymal stem cell‑derived exosomes (MSC‑Exos) have significant therapeutic potential for both KOA and OP. These exosomes enhance chondrocyte proliferation, modulate cartilage matrix synthesis and degradation, and suppress synovial inflammation, suggesting a novel therapeutic approach for KOA. However, their OP mechanisms remain unclear but may involve disrupted bone metabolic signaling, amplified inflammation, and dysregulated intercellular communication in the bone microenvironment. The present review summarizes MSC‑Exos research advances in KOA and OP, providing a foundation for future studies and clinical applications.
Keywords: exosomes; knee osteoarthritis; mesenchymal stem cells; osteoporosis.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Adipose Mesenchymal Stem Cells Derived Exosomes Ameliorates KOA Cartilage Damage and Inflammation by Activation of PINK1-Mediated Mitochondrial Autophagy.FASEB J. 2025 Jul 31;39(14):e70811. doi: 10.1096/fj.202501185R. FASEB J. 2025. PMID: 40641285
-
Injection of human umbilical cord mesenchymal stem cells exosomes for the treatment of knee osteoarthritis: from preclinical to clinical research.J Transl Med. 2025 Jun 11;23(1):641. doi: 10.1186/s12967-025-06623-y. J Transl Med. 2025. PMID: 40500748 Free PMC article. Clinical Trial.
-
Mesenchymal stem cells and knee osteoarthritis: A bibliometric analysis.Medicine (Baltimore). 2025 Jul 4;104(27):e43201. doi: 10.1097/MD.0000000000043201. Medicine (Baltimore). 2025. PMID: 40629584 Free PMC article.
-
Decadal analysis of efficacy and safety profiles of mesenchymal stem cells from varied sources in knee osteoarthritis patients: A systematic review and network meta-analysis.Exp Gerontol. 2024 Jul;192:112460. doi: 10.1016/j.exger.2024.112460. Epub 2024 May 20. Exp Gerontol. 2024. PMID: 38772192
-
Efficacy of adipose-derived stem cells and stromal vascular fraction for pain relief in Kellgren-Lawrence grade II-III knee osteoarthritis: A systematic review (2019-2024).J Orthop. 2025 Mar 21;70:95-106. doi: 10.1016/j.jor.2025.03.029. eCollection 2025 Dec. J Orthop. 2025. PMID: 40236276 Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous