

A comprehensive integrated disease management program for phenylketonuria (IDMP-PKU) from Türkiye: rationale, design and patient characteristics
- PMID: 40751258
- PMCID: PMC12317577
- DOI: 10.1186/s13023-025-03702-7
A comprehensive integrated disease management program for phenylketonuria (IDMP-PKU) from Türkiye: rationale, design and patient characteristics
Abstract
Background: Phenylketonuria is an autosomal recessive disorder characterized by the deficiency of phenylalanine hydroxylase, which converts phenylalanine into tyrosine. Diagnosis and prompt initiation of appropriate treatment shortly after birth are important for achieving optimal outcomes in phenylketonuria. IDMP-PKU is an ongoing study to gain insight into the patient journey and identify the unmet needs and areas for improvement in diagnosis, treatment, and follow-up of PKU in Türkiye.
Aim: To present the rationale and design of the IDMP-PKU study, as well as the findings from an interim analysis, describing baseline demographic, diagnosis, family history, and genetic testing data for 1553 children enrolled in the study.
Method: This is a multicenter, observational registry-based study, conducted in 3 tertiary pediatric metabolic clinics in Türkiye. The study provides a descriptive analysis of baseline demographic, diagnosis, family history, and genetic testing data of study population.
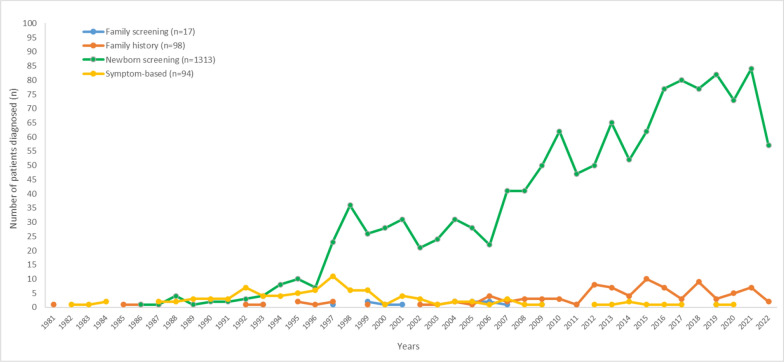
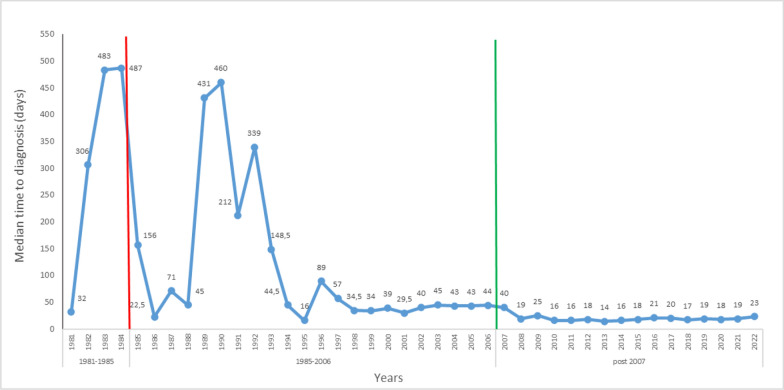
Results: The study included 1,553 patients (median age: 10 (IQR 5-18) years; 37.1% classical PKU) from 90% of the cities in Türkiye, diagnosed between 1981 and 2022. Parental consanguinity was reported in 43.5% of families (27.1% first cousins). The most frequently detected allelic variant was c.1066-11G > A (IVS-10-11G > A) (22.8%). Homozygous mutations were more common in patients with parental consanguinity (76.8% vs 17.1%; p < 0.001). The median time to diagnosis improved to 21 days after the implementation of the national newborn screening (NBS) program in December 2006 but 28.6% of patients were diagnosed after one month of age. Low level of maternal education was associated with longer time to diagnosis (p < 0.001).
Conclusions: Implementation of national NBS has contributed to earlier identification of patients with PKU. Increasing the number of screening laboratories and pediatric metabolic clinics will speed up the diagnostic process and help achieve the guideline-recommended time for diagnosis and initiation of treatment. In countries with high rates of consanguineous marriages, increasing public awareness of PKU and genetic counselling before marriage will be valuable in reducing the prevalence of PKU.
Keywords: Allelic variant; Consanguinity; Diagnosis; Maternal education; Newborn screening; Phenylketonuria.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Cukurova University, Faculty of Medicine, Clinical Research Ethics Committee approved the study (trial code: MON562.328.2) and related documents (19.11.2020–137/8). The protocol amendment for inclusion of SCL-90 and KBIT-2 tests as additional neurocognitive assessments, was also approved by the Ethics Committee (12.05.2022 -168/11).BU SONUÇLARA YER VERİLMEDİ AMA ANA ÇALIŞMA İLE İLİGLİ OLDUĞUNDNA KALMALI DİYE DÜŞÜNÜYORUM, The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines. Written informed consent was obtained from all volunteers or their legal guardians after the study procedures were explained. Consent for publication: Not applicable. Competing interests: The authors confirm that they do not have any potential conflict of interest. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Figures
References
-
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376(9750):1417–27. - PubMed
-
- PAHvdb: Phenylalanine Hydroxylase Gene Locus-Specific Database [Available from: http://www.biopku.org/home/pah.asp.
-
- Stone WL BH, Los E. Phenylketonuria: Treasure Island (FL): StatPearls Publishing; 2024 [StatPearls]. Available from: www.ncbi.nlm.nih.gov/books/NBK535378/.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
