

ARSA Variants Associated With Cognitive Decline and Long-Term Preservation of Motor Function in Metachromatic Leukodystrophy
- PMID: 40751594
- PMCID: PMC12317651
- DOI: 10.1002/jimd.70072
ARSA Variants Associated With Cognitive Decline and Long-Term Preservation of Motor Function in Metachromatic Leukodystrophy
Abstract
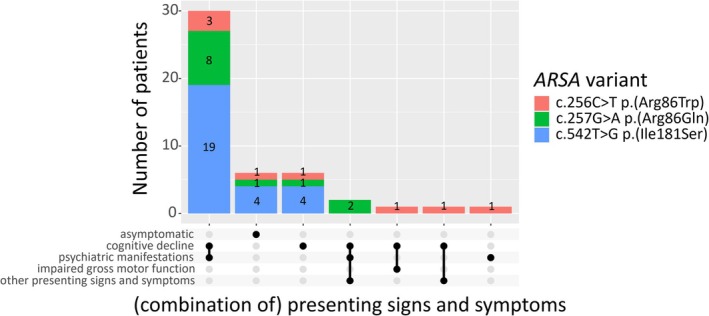
Patients with metachromatic leukodystrophy (MLD) show variable motor and cognitive decline. The ARSA variants c.256C>T, p.(Arg86Trp), c.257G>A, p.(Arg86Gln) and c.542T>G, p.(Ile181Ser) are associated with predominantly cognitive decline. This multinational study analyzed MLD onset type, presenting signs/symptoms, cognitive function, gross motor function, central motor tract involvement, MRI severity score, peripheral neuropathy, and survival of 47 patients (three homozygous for c.256C>T and five, twelve and 27 compound heterozygous for c.256C>T, c.257G>A, or c.542T>G and another ARSA variant, respectively). Eleven underwent hematopoietic stem cell transplantation (HSCT). Onset was late-juvenile (46.8%) or adult (44.7%) with predominantly cognitive decline (n = 40/41 symptomatic patients). At diagnosis, untreated patients typically retained independent walking (100%), sparing of central motor tracts (87.5%), and absence of demyelinating neuropathy (95.5%), which persisted in follow-up for most (76.5%, 71.4%, and 64.7%, respectively). Early-juvenile onset and rapid motor decline occurred only in patients compound heterozygous for c.256C>T and a severe second variant (n = 4), showing central motor tract involvement at diagnosis. One untreated and one treated patient died of disease progression, and another from HSCT complications. All other treated patients retained independent walking, and four of five tested normal cognitive function. Median MRI severity score remained lower in treated (13) than untreated patients (25). The phenotype of c.256C>T carriers depends on the severity of the second ARSA variant. Patients harboring c.257G>A or c.542T>G show late-juvenile or adult onset with cognitive decline and preserved motor function, usually associated with sparing of central motor tracts. In these patients, cognitive function and MRI severity score should be preferred treatment outcomes.
Keywords: ARSA gene; arylsulfatase A; genetic association studies; hematopoietic stem cell transplantation; metachromatic leukodystrophy.
© 2025 The Author(s). Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
Nicole I. Wolf is advisor and/or co‐investigator for trials in MLD (Shire/Takeda, Orchard, Evidera) and other leukodystrophies (Ionis, PassageBio, Vigil Neuro), with all payments to the institution. Annette Bley is coinvestigator/advisor for trials in MLD (Shire/Takeda, Orchard). Caroline Sevin is an advisor and/or investigator for trials in MLD (Shire/Takeda, Orchard). Francesca Fumagalli is an investigator of gene therapy clinical trials for MLD sponsored by Orchard Therapeutics, the license holder of Libmeldy, and acts as a consultant for the ad hoc Advisory Board of Orchard Therapeutics and Takeda. Carla E. M. Hollak is involved in premarketing studies with Sanofi, Protalix, and Idorsia, for which financial arrangements are made with Amsterdam UMC Research BV. These activities are unrelated to the content of this manuscript. Ludger Schöls served as a consultant for Vico Therapeutics, Vigil Neuroscience, and Novartis unrelated to MLD and the manuscript. Samuel Groeschel received an Institutional Research Support from Shire plc and Orchard, and is an advisor and coinvestigator for trials in MLD (Shire/Takeda, Orchard) without personal payments. Yann Nadjar received a research grant from Orchard. The other authors declare no conflicts of interest.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
