

Enlarged PML-nuclear bodies trigger conflicting cell cycle signal-mediated cytotoxicity in leukemia cells
- PMID: 40753178
- PMCID: PMC12318136
- DOI: 10.1038/s41419-025-07911-7
Enlarged PML-nuclear bodies trigger conflicting cell cycle signal-mediated cytotoxicity in leukemia cells
Abstract
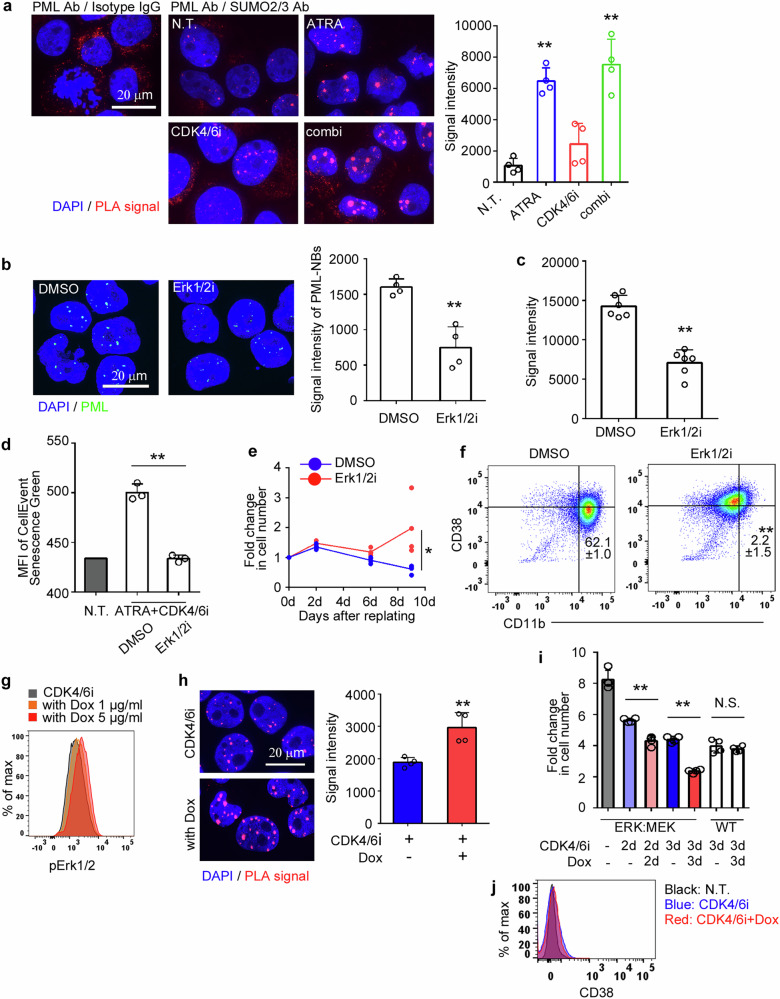
Accumulating evidence suggests that mitogenic signaling during cell cycle arrest can lead to severe cytotoxic outcomes, such as senescence, though the underlying mechanisms remain poorly understood. Here, we explored the link between cell cycle dynamics and the formation of PML-nuclear bodies (PML-NBs), intranuclear structures known to mediate cellular stress responses. Our findings demonstrate that PML-NBs increase their number during interphase arrest. Moreover, the activation of mitogenic ERK signaling by all-trans retinoic acid (ATRA) during CDK4/6 inhibitor-induced cell cycle arrest synergistically enhances the formation of larger PML-NBs by associating with SUMO. This enlargement, triggered by the simultaneous engagement of opposing cell cycle signals, leads to potent cytotoxicity accompanied by either terminal differentiation or apoptosis, depending on the cell type, across multiple acute myeloid leukemia (AML) cell lines. Importantly, in an AML mouse model, this combination treatment significantly improved therapeutic efficacy with minimal effects on normal hematopoiesis. Our results introduce conflicting cell cycle signal-induced cytotoxicity as a promising therapeutic strategy for AML.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethics approval and consent to participate: All methods were performed in accordance with the relevant guidelines and regulations. The animal studies were approved by the Committee of Laboratory Animals of Kanazawa University (AP-214222). No studies including human subjects were performed in this manuscript.
Figures





Similar articles
-
Microvesicle inhibition enhances the therapeutic effects of ATRA in acute promyelocytic leukemia cells via changes in miRNAs: the promising antileukemic potential of imipramine.Clin Exp Med. 2025 Jun 25;25(1):217. doi: 10.1007/s10238-025-01763-3. Clin Exp Med. 2025. PMID: 40555813 Free PMC article.
-
Duocarmycin SA Reduces Proliferation and Increases Apoptosis in Acute Myeloid Leukemia Cells In Vitro.Int J Mol Sci. 2024 Apr 14;25(8):4342. doi: 10.3390/ijms25084342. Int J Mol Sci. 2024. PMID: 38673926 Free PMC article.
-
Perturbing LSD1 and WNT rewires transcription to synergistically induce AML differentiation.Nature. 2025 Jun;642(8067):508-518. doi: 10.1038/s41586-025-08915-1. Epub 2025 Apr 16. Nature. 2025. PMID: 40240608 Free PMC article.
-
The Dynamic Regulation of Daxx-Mediated Transcriptional Inhibition by SUMO and PML NBs.Int J Mol Sci. 2025 Jul 12;26(14):6703. doi: 10.3390/ijms26146703. Int J Mol Sci. 2025. PMID: 40724953 Free PMC article. Review.
-
Thrombopoietin mimetics for patients with myelodysplastic syndromes.Cochrane Database Syst Rev. 2017 Sep 30;9(9):CD009883. doi: 10.1002/14651858.CD009883.pub2. Cochrane Database Syst Rev. 2017. PMID: 28962071 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
