

Identification of prognostic genes related to T cell proliferation in papillary thyroid cancer based on single-cell RNA sequencing and bulk RNA sequencing data
- PMID: 40753315
- PMCID: PMC12317885
- DOI: 10.1007/s10238-025-01826-5
Identification of prognostic genes related to T cell proliferation in papillary thyroid cancer based on single-cell RNA sequencing and bulk RNA sequencing data
Abstract
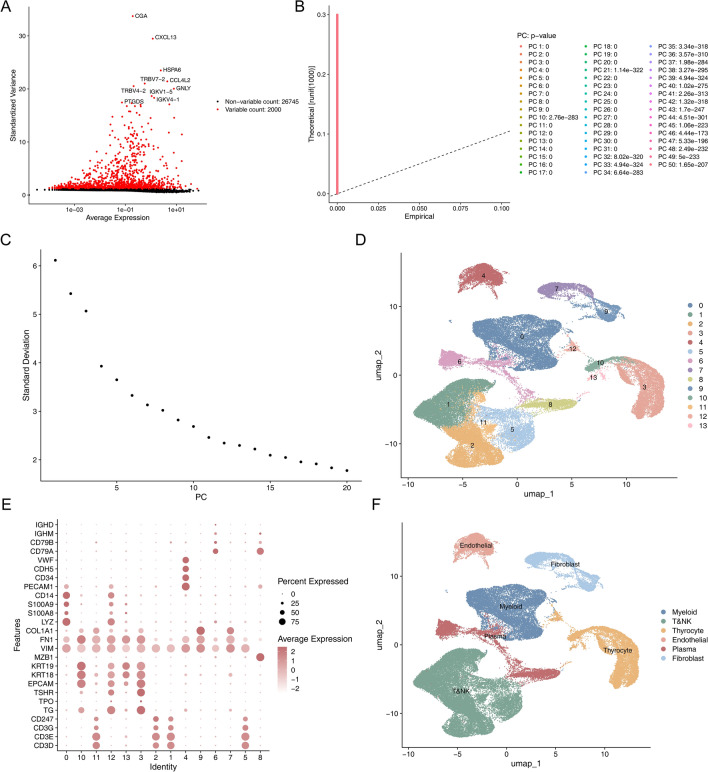
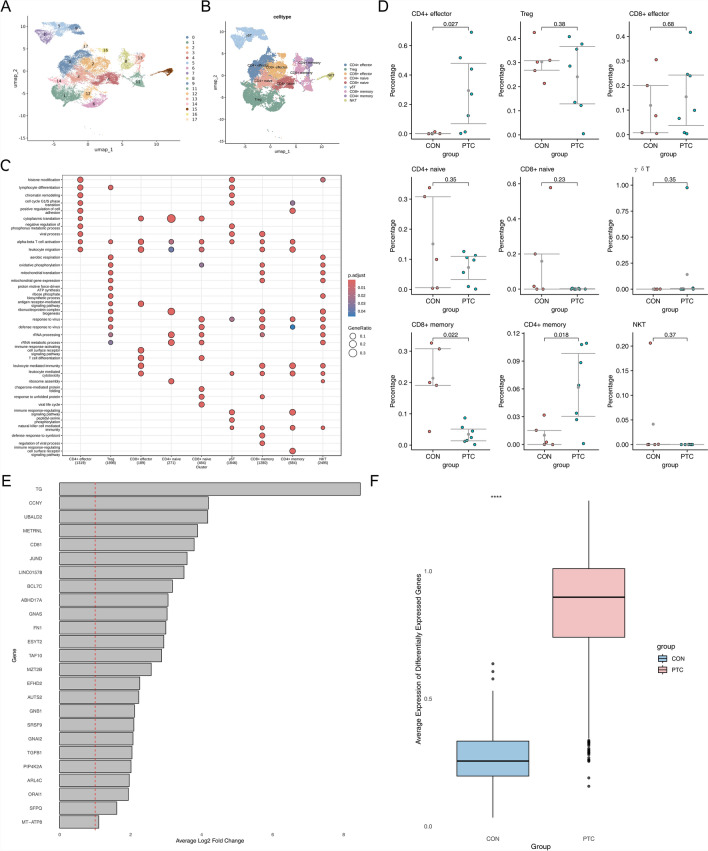
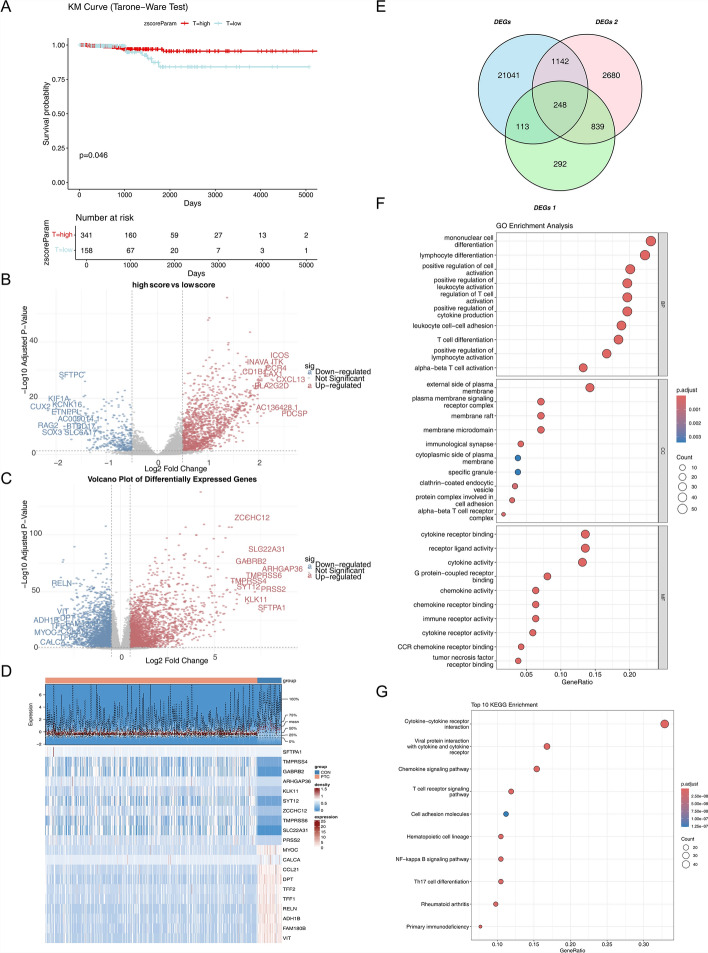
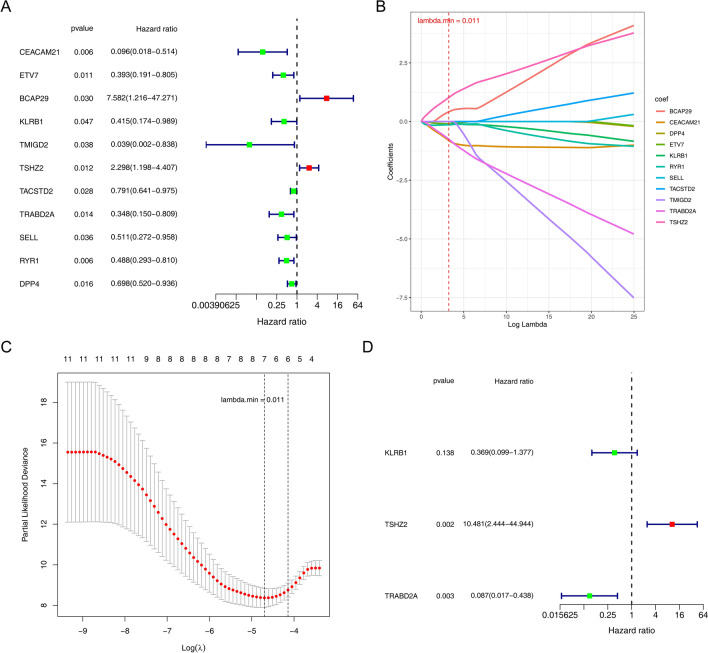
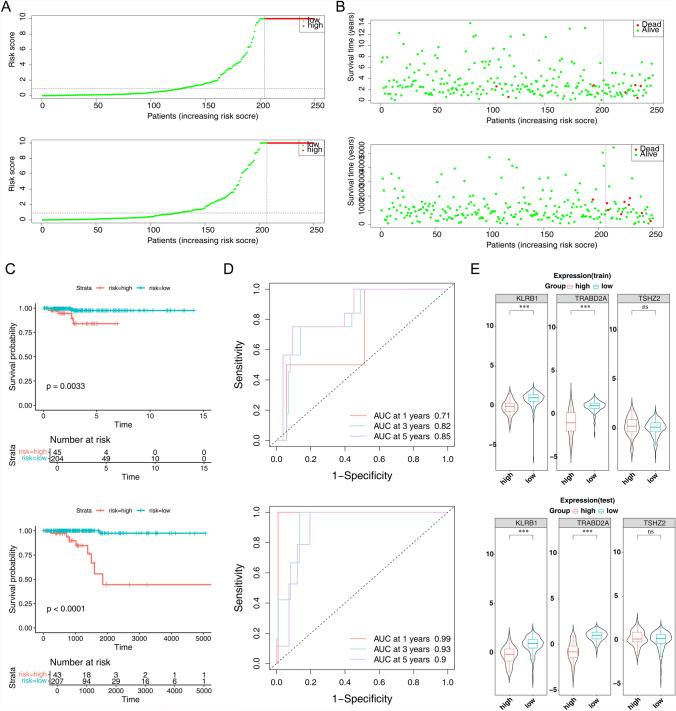
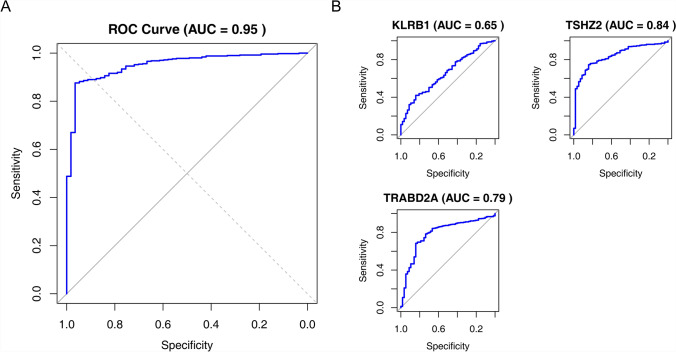
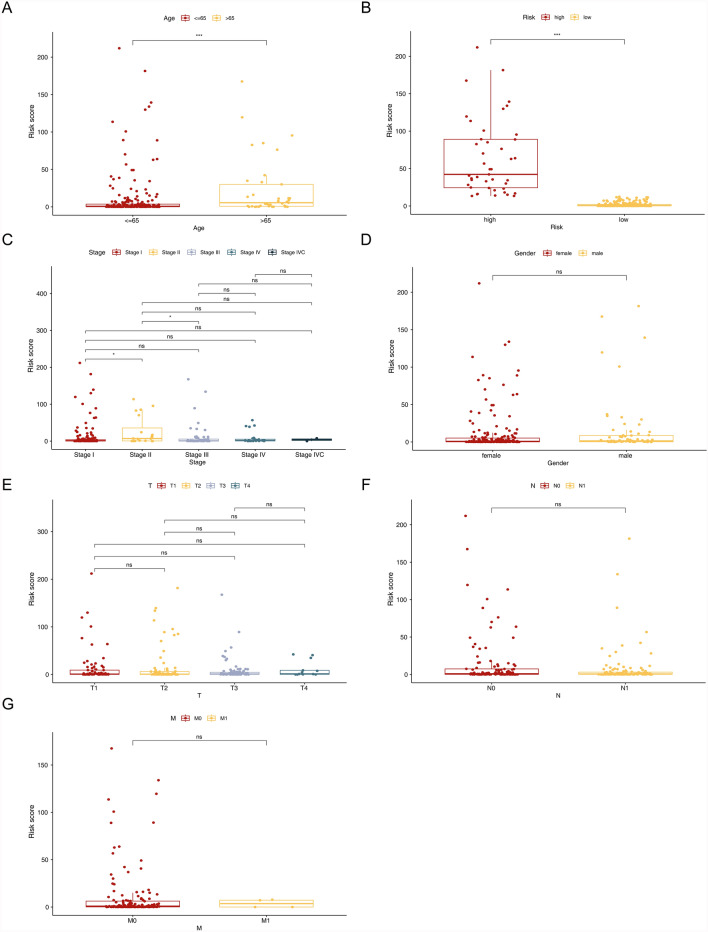
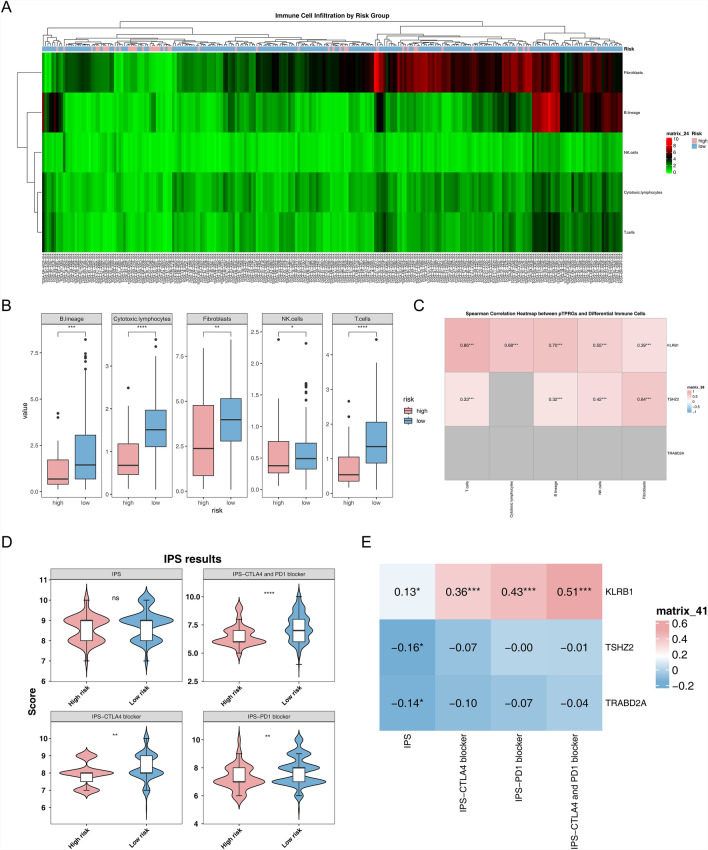
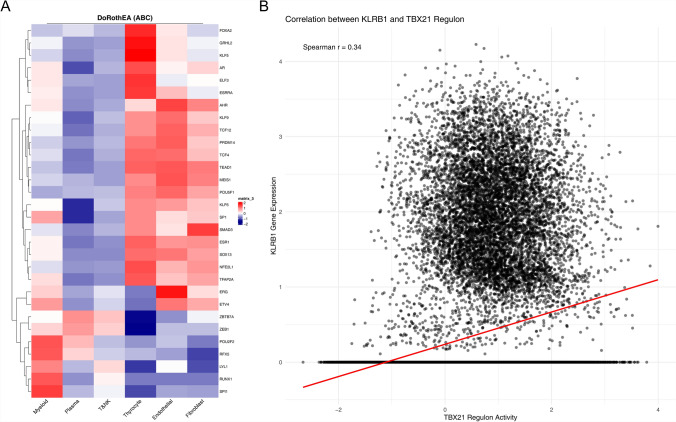
Papillary thyroid carcinoma (PTC) is the main pathological subtype of thyroid cancer. Given the strong association between T cells and PTC, this study focused on the prognostic value and potential molecular mechanisms of T cell proliferation-related genes (TPRGs) in PTC. ScRNA-seq data were analyzed to identify key cells and subpopulations based on public databases. Candidate genes were determined by intersecting differentially expressed genes (DEGs) from differential expression analysis of key subpopulations, high- and low-TPRGs score groups, and bulk RNA-seq data. Prognostic genes were then determined via Cox regression and a machine learning algorithm. A risk model was formed. PTC patients were grouped into high-risk and low-risk groups by risk score. Subsequently, the immune microenvironment was analyzed. Finally, cell communication analysis and pseudotime analysis were accomplished. T/NK cells were selected as key cells. Moreover, CD4+ memory cells were selected as the key subpopulation. Meanwhile, KLRB1, TSHZ2, and TRABD2A were spotted as prognostic genes. The risk model had better prognostic value. Additionally, 5 DICs were spotted in two risk groups. Besides, the scores of IPS-CTLA4 and PD-L1 blocker, IPS-CTLA4 blocker, and IPS-PD-1 blocker were lower in the HRG group. In addition, the most substantial receptor-ligand interaction in T/NK cells was found to be CLEC2C-KLRB1. Moreover, as T/NK cells differentiated, the expression level of KLRB1 was observed to rise slowly at first and then decline rapidly. KLRB1, TSHZ2, and TRABD2A were spotted as prognostic genes, providing new PTC prognosis and treatment strategies.
Keywords: Immune microenvironment; Papillary thyroid cancer; Prognostic genes; T cell proliferation.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Conflicts of interest: The authors declare no competing interests. Ethics approval: Not applicable. Consent to participate: Not applicable. Consent to publish: Not applicable.
Figures
Similar articles
-
Single-cell RNA sequencing and multi-omics analysis of prognosis-related staging in papillary thyroid cancer.Cancer Immunol Immunother. 2025 Jul 12;74(8):267. doi: 10.1007/s00262-025-04101-4. Cancer Immunol Immunother. 2025. PMID: 40650680 Free PMC article.
-
Molecular function validation and prognostic value analysis of the cuproptosis-related gene ferredoxin 1 in papillary thyroid carcinoma.Sci Rep. 2025 Jul 23;15(1):26845. doi: 10.1038/s41598-025-11151-2. Sci Rep. 2025. PMID: 40702045 Free PMC article.
-
Bioinformatics identification and validation of m6A/m1A/m5C/m7G/ac4 C-modified genes in oral squamous cell carcinoma.BMC Cancer. 2025 Jul 1;25(1):1055. doi: 10.1186/s12885-025-14216-7. BMC Cancer. 2025. PMID: 40597017 Free PMC article.
-
The Association between Lymphocytic Thyroiditis and Papillary Thyroid Cancer Harboring Mutant BRAF: A Systematic Review and Meta-Analysis.Thyroid. 2024 Sep;34(9):1082-1093. doi: 10.1089/thy.2024.0142. Epub 2024 Jul 18. Thyroid. 2024. PMID: 38940753
-
Systemic treatments for metastatic cutaneous melanoma.Cochrane Database Syst Rev. 2018 Feb 6;2(2):CD011123. doi: 10.1002/14651858.CD011123.pub2. Cochrane Database Syst Rev. 2018. PMID: 29405038 Free PMC article.
References
-
- Shaerf DA, Roberton JB, Horwitz MD. Traumatic pseudoaneurysm of the ulnar artery treated with ultrasound guided thrombin injection. J Hand Surg (European Vol). 2019;44(6):652–4. - PubMed
-
- McLeod DSA, et al. Contemporary debates in adult papillary thyroid cancer management. Endocr Rev. 2019;40(6):1481–99. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
