

Insights into diversity, host range, and evolution of iflaviruses in Lepidoptera through transcriptome mining
- PMID: 40755814
- PMCID: PMC12315680
- DOI: 10.1093/ve/veaf051
Insights into diversity, host range, and evolution of iflaviruses in Lepidoptera through transcriptome mining
Abstract
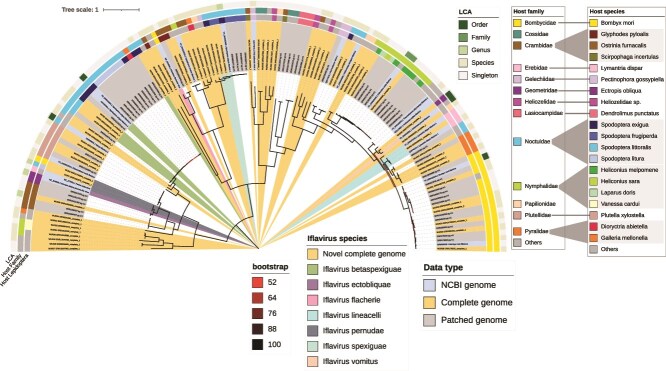
Insects are associated with a wide variety of diverse RNA viruses, including iflaviruses, a group of positive stranded RNA viruses that mainly infect arthropods. Whereas some iflaviruses cause severe diseases in insects, numerous iflaviruses detected in healthy populations of butterflies and moths (order: Lepidoptera) do not show apparent symptoms. Compared to other hosts, only few iflavirus genomes for lepidopteran hosts could be found in publicly available databases and we know little about the occurrence of iflaviruses in natural and laboratory lepidopteran populations. To expand the known diversity of iflaviruses in Lepidoptera, we developed a pipeline to automatically reconstruct virus genomes from public transcriptome data. We reconstructed 1548 virus genomes from 55 different lepidopteran species, which were identified as coding-complete based on their length. To include incompletely assembled genomes, we developed a reference-based patching approach, resulting in 240 patched genomes. By including publicly available genomes, we inferred a phylogeny consisting of 139 non-redundant iflavirus genomes. Of these, 65 represent novel complete genomes, of which 39 might even belong to novel virus species. Our analysis expanded virus host range, where highly similar viruses were found in the transcriptomes of different lepidopteran species, genera, or even families. Additionally, we find two groups of lepidopteran species depending on the diversity of viruses that infect them: some species were only infected by closely related viruses, whereas other species are infected by highly diverse viruses from different regions of the phylogeny. Finally, we show that the evolution of one virus species, Iflavirus betaspexiguae, is impacted by recombination within the species, which is also supported by the co-occurrence of multiple strains within the data sets. Our analysis demonstrates how data mining of publicly available sequencing data can be used at a large scale to reconstruct intra-family viral diversity which serves as a basis to study virus host range and evolution. Our results contain numerous novel viruses and novel virus-host associations, including viruses for relevant insect pests, highlighting the impact of iflaviruses in insect ecology and as potential biological control agents in the future.
Keywords: data mining; iflavirus; phylogeny.
© The Author(s) 2025. Published by Oxford University Press.
Figures



References
-
- Barrera G, Simón O, Villamizar L et al. Spodoptera frugiperda multiple nucleopolyhedrovirus as a potential biological insecticide: genetic and phenotypic comparison of field isolates from Colombia. Biol Control 2011;58:113–20. 10.1016/j.biocontrol.2011.04.009 - DOI
LinkOut - more resources
Full Text Sources
