

Direct whole-genome sequencing enables strain typing of unculturable Neisseria meningitidis from oropharyngeal carriage specimens
- PMID: 40758406
- PMCID: PMC12321007
- DOI: 10.1099/mgen.0.001464
Direct whole-genome sequencing enables strain typing of unculturable Neisseria meningitidis from oropharyngeal carriage specimens
Abstract
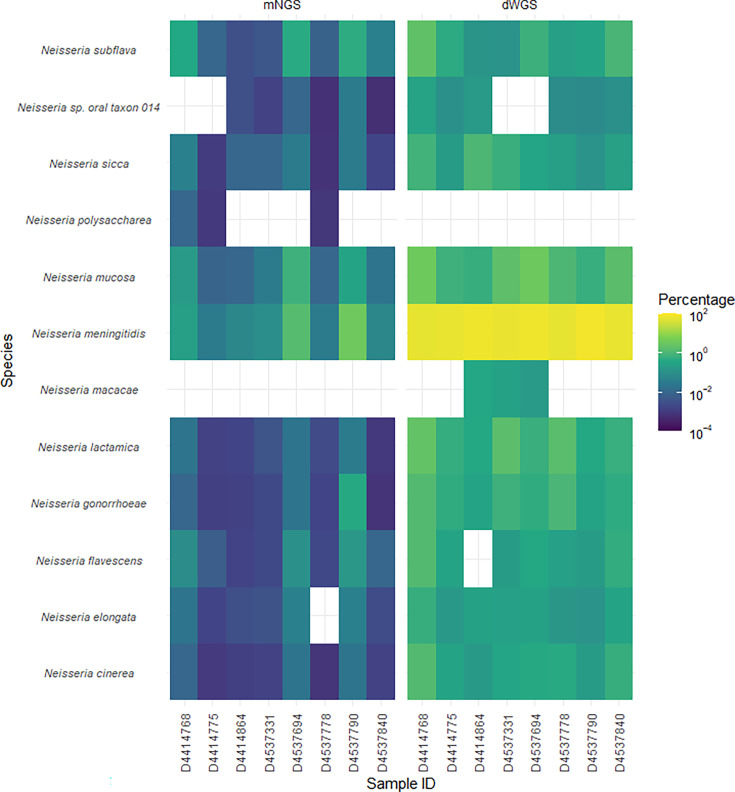
Oropharyngeal carriage of Neisseria meningitidis (N.m.) is a prerequisite for invasive meningococcal disease. As such, genomic surveillance of disease-causing carriage strains can inform targeted public health responses. However, whole-genome sequencing (WGS) from isolates is often precluded due to the high rates of culture failure for N.m. samples collected in carriage studies. This study outlines an alternative method to sequence N.m. directly from oropharyngeal specimens that enables high-resolution molecular fine typing.We performed direct probe-capture enrichment WGS (dWGS) of N.m. on oropharyngeal specimens from the 'B part of it' South Australian and 'B part of it NT' Northern Territory meningococcal carriage studies (NCT03089086 and NCT04398849). Sequences were analysed using currently available bioinformatic tools, including the characterization of genogroup, multi-locus sequence typing (MLST), Bexsero Antigen Sequence Typing (BAST), porA and fetA type.Sensitivity of dWGS typing compared to WGS for genogroup, MLST, porA, fetA and BAST schemes was 88.89%, 72.22%, 100%, 94.44% and 88.24%, respectively. Genogroup and porA type were more reliably characterized in unculturable samples compared to the other typing schemes assessed. Factors that influenced accurate fine typing included the amount and proportion of N.m. sequences, and the proportion of other Neisseria species in enriched sequencing libraries. An alternative phylogenetic method (phylotyping) correctly predicted the clonal complex for 93.46% of the samples assessed. These results demonstrate that dWGS enables high-resolution molecular fine typing and can be applied to unculturable samples in N.m. carriage studies.
Keywords: typing; Neisseria meningitidis; metagenomics; whole-genome sequencing.
Conflict of interest statement
H.M. is an investigator on vaccine trials sponsored by GSK, Novavax, Sanofi Pasteur and Pfizer. H.M.’s, M.Mc.’s and A.L.’s institution receives funding for investigator-led studies from Pfizer and GSK. H.M., M.Mc. and A.L. receive no personal payments from industry.
Figures








References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials