

GenomicLayers: sequence-based simulation of epi-genomes
- PMID: 40759911
- PMCID: PMC12323044
- DOI: 10.1186/s12859-025-06224-y
GenomicLayers: sequence-based simulation of epi-genomes
Abstract
Background: Cellular development and differentiation in Eukaryotes depends upon sequential gene regulatory decisions that allow a single genome to encode many hundreds of distinct cellular phenotypes. Decisions are stored in the regulatory state of each cell, an important part of which is the epi-genome-the collection of proteins, RNA and their specific associations with the genome. Additionally, further cellular responses are, in part, determined by this regulatory state. To date, models of regulatory state have failed to include the contingency of incoming regulatory signals on the current epi-genetic state and none have done so at the whole-genome level.
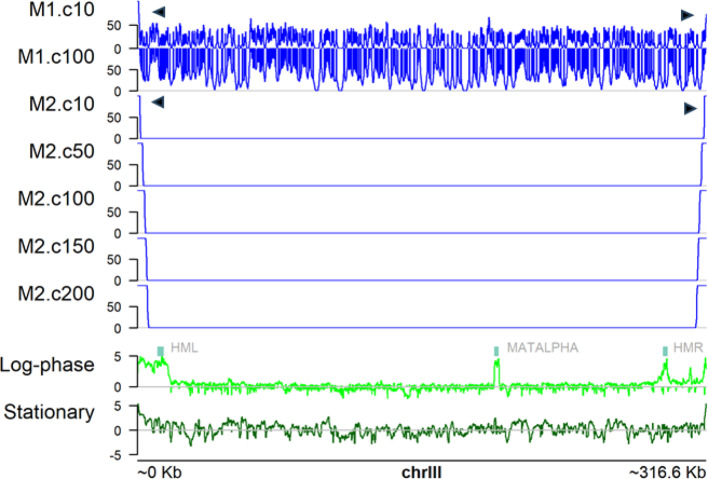
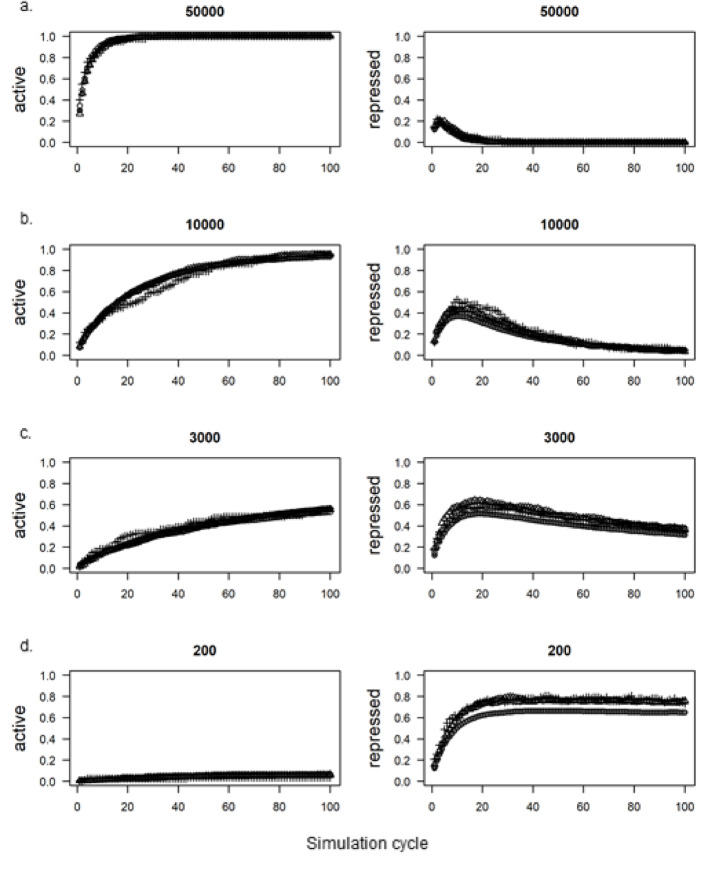
Results: Here we introduce GenomicLayers, a new R package to run rules-based simulations of epigenetic state changes genome-wide in Eukaryotes. Simulations model the accumulation of changes to genome-wide layers by user-specified binding factors. As a first exemplar, we show two versions of a simple model of the recruitment and spreading of epigenetic marks near telomeres in the yeast Saccharomyces cerevisiae. By combining the output from 100 runs of the simulation, we generate whole genome predictions of epigenetic state at 1 bp resolution. The example yeast models are included within a 'vignette' with the GenomicLayers package, which is available at https://github.com/davetgerrard/GenomicLayers . To demonstrate the use of GenomicLayers on the full human reference genome (hg38), we show the results from parameter refinement on a simplistic model of the action of pluripotency factors against a self-spreading repressor seeded at CpG islands. The human genome model is included in supplementary information as an R script.
Conclusions: GenomicLayers enables scientists working on diverse eukaryotic organisms to test models of gene regulation in silico. Applications include epigenetic silencing, activation by combinatorial binding of transcription factors and the sink effects caused by down-regulation of components of epigenetic regulators. The software is intended to be used to parameterise, refine and combine models and thereby capitalise on data from the thousands of studies of Eukaryotic epigenomes.
Keywords: Development; Epigenome; Genome; R; Simulation.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not applicable. Consent for publication: Not applicable. Competing interests: The authors declare that they have no competing interests.
Figures

References
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
